

Erectile dysfunction (ED) remains one of the most prevalent and psychologically burdensome male health disorders, affecting approximately one in five men over the age of forty. Although vasculature, neuroregulation, endocrine balance, and psychosocial factors all contribute critically to erectile performance, recent scientific attention has pivoted toward an often-underestimated protagonist: the cavernous smooth muscle cell (CSMC). These contractile cells—strategically embedded within the corpus cavernosum—govern hemodynamic transitions that define erection quality, rigidity, duration, and vascular occlusion.
The review underlying this article explores CSMCs not merely as passive contractile elements but as dynamic regulators of nitric oxide (NO) signaling, calcium homeostasis, redox balance, ion channel activity, and apoptosis. Images and diagrams within the PDF (e.g., physiological pathway schematic on page 3) illustrate the intricate choreography of neurotransmitters, ion fluxes, and endothelial mediators required for a sustained erection.
In this article, we revisit these mechanisms through a translational lens, explaining why smooth muscle dysfunction—not aging per se—may represent the earliest and most modifiable driver of erectile decline. By exploring calcium dynamics, oxidative stress, apoptosis, endothelial–muscular cross-talk, and the role of molecular signaling cascades, we aim to clarify why CSMCs sit at the center of both erectile physiology and pathology.
The Physiology of Erection: A Story Written by Smooth Muscle Relaxation
An erection begins not with blood flow, but with smooth muscle relaxation—a distinction that shifts emphasis from the vasculature to the intracellular machinery of CSMCs. As described in the PDF’s introductory physiology section , erection is essentially a hemodynamic switch triggered by neurotransmitters such as NO, prostaglandins, vasoactive intestinal peptide, and endothelial hyperpolarizing factors. These mediators reduce CSMC tone, enabling rapid expansion of sinusoidal spaces, venous occlusion, and the sharp rise in intracavernosal pressure.
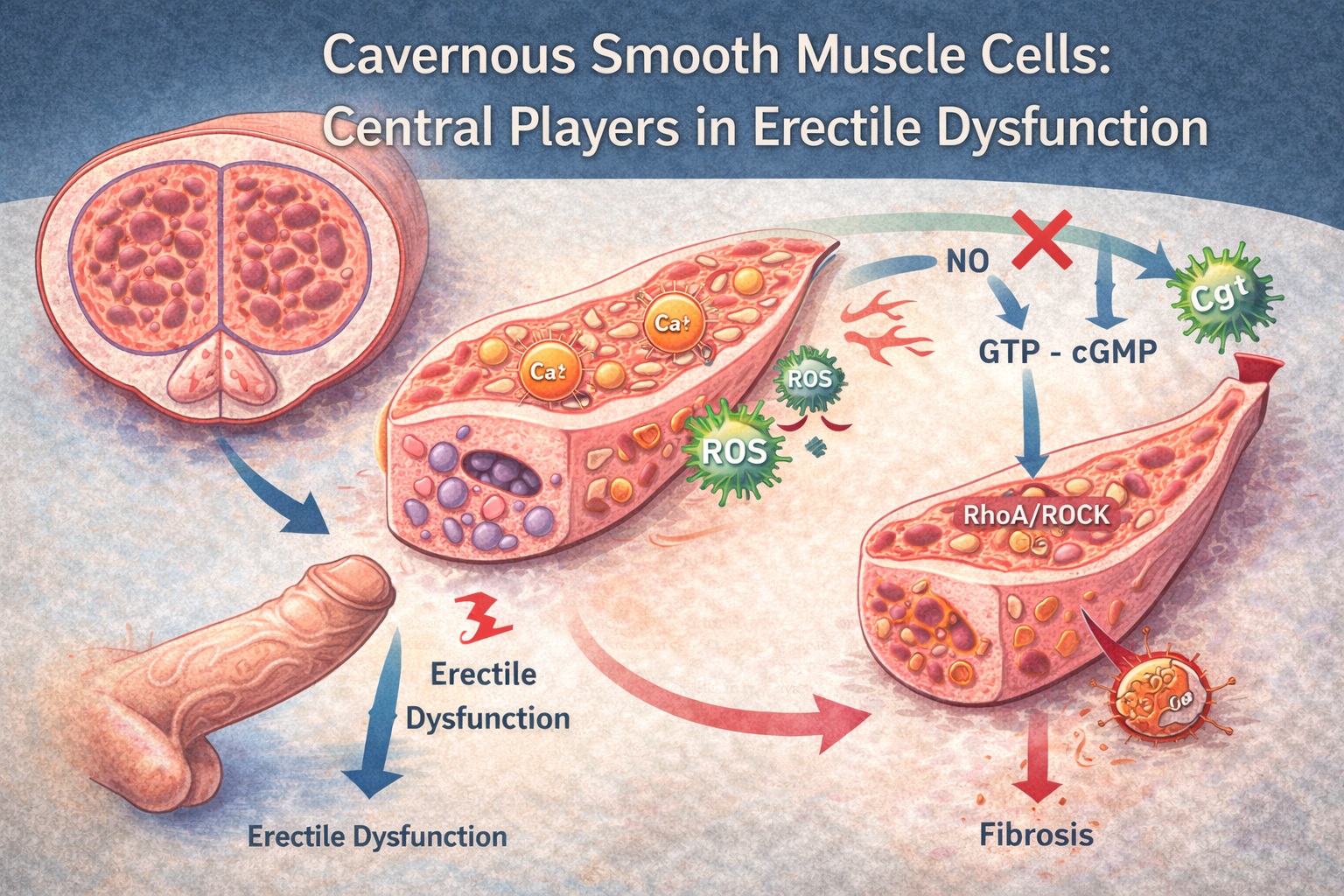
Smooth muscle relaxation is governed primarily by NO–cGMP signaling. Upon sexual stimulation, neuronal and endothelial nitric oxide synthase (nNOS and eNOS) produce NO, which diffuses into CSMCs and activates soluble guanylate cyclase (sGC). This increases cGMP levels, engaging protein kinase G (PKG), which in turn reduces intracellular calcium concentrations while opening potassium channels. As depicted on page 4 of the PDF, the result is hyperpolarization, reduced contractility, and controlled vasodilation.
Normal CSMCs maintain a delicate equilibrium between contractile and synthetic phenotypes. While the contractile phenotype preserves firmness and vascular control, the synthetic phenotype emerges during pathological remodeling—promoting fibrosis, apoptosis, and oxidative stress. Unfortunately, ED often represents a shift from contractile to synthetic behavior, disrupting the capacity for proper relaxation.
One critical aspect is the dynamic regulation of calcium. Elevated Ca²⁺ levels promote contraction, whereas reductions in Ca²⁺ or its sensitivity favor relaxation. In ED, CSMCs lose their ability to regulate intracellular Ca²⁺ effectively. Channels such as L-type Ca²⁺ channels, store-operated Ca²⁺ entry channels, and ryanodine receptors become dysregulated, making muscle cells refractory to neurotransmitter-induced relaxation. With time, this impaired Ca²⁺ handling evolves from a functional disturbance into structural remodeling.
Thus, erection is essentially a tale of calcium, cyclic nucleotides, and the smooth muscle’s willingness to relax. When the muscle becomes stiff—biochemically or structurally—the entire system falters.
Calcium Homeostasis: The Double-Edged Sword of Contractility
The study emphasizes calcium’s central role in CSMC physiology, as detailed in its biochemical and mechanistic sections . Proper erectile function requires a finely tuned balance between Ca²⁺ influx, release from intracellular stores, sequestration, and efflux. Disruption at any point leads to impaired relaxation.
At baseline, cavernosal smooth muscle contraction is driven by myosin light-chain kinase (MLCK), activated by Ca²⁺–calmodulin complexes. During sexual stimulation, PKG phosphorylates and inhibits MLCK, reducing Ca²⁺ sensitivity. Simultaneously, PKG enhances myosin light-chain phosphatase (MLCP), promoting dephosphorylation and relaxation.
In ED, however, oxidative stress disrupts this process. As shown in the oxidative stress diagram on page 6, reactive oxygen species (ROS) increase Ca²⁺ channel opening, inhibit PKG activity, and impair NO bioavailability. This “oxidized muscle state” makes relaxation nearly impossible, no matter how much NO is liberated upstream.
Another challenge is apoptosis-driven calcium dysregulation. Apoptotic signaling damages sarcoplasmic reticulum (SR) proteins, leading to uncontrolled Ca²⁺ leakage. Over time, chronic calcium overload drives fibrosis, cellular atrophy, and further dysfunction. Reduced endothelial NO production exacerbates this, creating a feedback loop of oxidative stress and Ca²⁺ mishandling.
These calcium-dependent mechanisms explain why ED is not merely a vascular disease but a smooth muscle disease—one shaped by biochemical exhaustion and structural remodeling.
Oxidative Stress and Apoptosis: The Silent Saboteurs of Cavernosal Integrity
Oxidative stress remains one of the most widely accepted mechanisms underlying CSMC impairment. ROS levels increase with aging, diabetes, hypertension, smoking, obesity, and chronic inflammation. The PDF provides a detailed overview of ROS-driven pathology, including mitochondrial dysfunction and cellular apoptosis (see page 5, ROS pathway illustration) .
ROS neutralize NO, converting it to peroxynitrite—a potent oxidant that damages lipids, proteins, DNA, and endothelial integrity. This not only reduces NO bioavailability but also directly harms CSMC membranes and mitochondria. Mitochondrial depolarization leads to cytochrome c release, caspase activation, and apoptotic death of smooth muscle cells.
Apoptosis poses a particular threat to erectile physiology because CSMCs are not easily replaced. As the ratio of smooth muscle to collagen diminishes, the penis loses its compliance and becomes structurally incapable of trapping blood. This is not functional ED—it is architectural ED, a form that responds poorly even to PDE5 inhibitors.
The paper highlights conditions such as diabetes and cavernous nerve injury where apoptosis accelerates dramatically. Hyperglycemia-induced ROS amplify calcium overload, mitochondrial fragmentation, and endothelial dysfunction. Men recovering from radical prostatectomy face similar challenges due to denervation-triggered apoptosis, which begins within days after surgery.
Once apoptosis-driven fibrosis sets in, intracavernosal pressure cannot rise adequately, even when upstream signals remain intact. This explains why early ED interventions focus increasingly on preserving smooth muscle mass rather than simply augmenting hemodynamics.
Ion Channels: Subtle Regulators with Outsized Influence
Although ion channels might appear as mere technical details in penile physiology, their influence on CSMC behavior is profound. The review discusses various potassium channels (BKCa, Kir, KATP), chloride channels, and calcium channels that determine membrane potential and contractile readiness.
BKCa channels, for instance, respond to voltage and intracellular calcium, causing hyperpolarization that limits Ca²⁺ influx. When ROS block these channels—as shown in data summarized on page 7—the cell becomes chronically depolarized, inhibiting relaxation.
Similarly, TRP channels (transient receptor potential) mediate Ca²⁺ influx in response to mechanical and chemical stimuli. Upregulation of TRPC channels in diabetes and aging creates a pathological Ca²⁺ leak, maintaining the cell in a semi-contracted, dysfunctional state.
PDE5 inhibitors indirectly influence channel behavior by increasing cGMP and PKG activation, which enhances potassium channel opening. This explains why patients with channelopathies—especially diabetic men—often respond incompletely to PDE5 inhibitors: the channels themselves are structurally or functionally impaired.
Through these channel interactions, the study reinforces the concept that smooth muscle dysfunction is not monolithic but a convergence of membrane instability, redox imbalance, and disrupted calcium dynamics.
Signaling Pathways Driving Dysfunction: From NO Deficiency to RhoA/ROCK Activation
The article highlights several signaling pathways that contribute to ED progression, many of which act directly upon CSMC tone. In the pathway summaries on pages 5–8, the authors outline how various intracellular mechanisms interact .
One of the most important antagonists of relaxation is the RhoA/ROCK pathway, which increases Ca²⁺ sensitivity of contractile proteins. ROCK inhibits MLCP, preserving myosin phosphorylation even in the absence of high Ca²⁺ levels. In ED, chronic activation of RhoA/ROCK—particularly in conditions such as diabetes, hypertension, and endothelial dysfunction—creates a biochemical lock on smooth muscle tone.
Inflammatory cytokines, including TNF-α and IL-6, further activate RhoA/ROCK, contributing to fibrosis and impaired relaxation. This pathway becomes a bottleneck where even increased NO or pharmacologic PDE5 inhibition may not fully overcome contractile resistance.
Conversely, the cAMP/PKA pathway complements the cGMP/PKG axis by promoting relaxation through alternate phosphorylation targets. Prostaglandin-mediated cAMP production, illustrated in the diagram on page 4, acts synergistically with NO to suppress contractility. When oxidative stress reduces adenylate cyclase activity or increases PDE expression, this compensatory mechanism collapses.
Together, these pathways form a biochemical “tug-of-war.” In healthy tissue, relaxation pathways dominate effortlessly. In ED, contractile pathways gain the upper hand—until hemodynamic failure becomes clinically apparent.
Structural Remodeling: When Smooth Muscle Loss Becomes Irreversible
In later stages of ED, CSMC decline leads to stark architectural remodeling. Fibrosis replaces muscle; collagen and elastin disorganize; and the veno-occlusive mechanism collapses. As emphasized repeatedly throughout the paper, this remodeling is both a cause and effect of progressive dysfunction.
Histological images in the PDF (pages 8–9) demonstrate loss of smooth muscle density and increased deposition of collagen fibers—findings typical in diabetes, Peyronie’s disease, post-prostatectomy states, and chronic hypoxia. Once the smooth muscle-to-collagen ratio falls below a physiological threshold, PDE5 inhibitors lose effectiveness, as no amount of cGMP can relax tissue that has structurally ceased to behave like muscle.
This underscores the importance of early ED management focused on preventing apoptosis and fibrosis. Therapies under investigation—including stem cell injections, NO-donors, ROCK inhibitors, low-intensity shockwave therapy, and regenerative biologics—all target smooth muscle preservation or restoration.
Thus, ED management is shifting from hemodynamic rescue to tissue-level preservation, aligning with the study’s central message: the CSMC is not peripheral to erectile function—it is erectile function.
Conclusion
The article underlying this review makes a compelling argument that cavernous smooth muscle cells are the true central regulators of erectile physiology. Their ability to relax, resist oxidative injury, regulate calcium, and maintain structural integrity dictates whether neurovascular input will translate into a functional erection.
ED, therefore, is less a disorder of aging and more a disorder of cellular resilience. CSMCs deteriorate under oxidative stress, inflammation, endothelial dysfunction, apoptosis, and signaling imbalances—ultimately leading to fibrosis and irreversible mechanical failure.
Recognizing smooth muscle cells as the “core unit” of erectile health reframes ED treatment around prevention, preservation, and cellular restoration—not merely symptomatic vasodilation.
FAQ
1. Why are cavernous smooth muscle cells so important in erectile dysfunction?
Because erection depends on their relaxation. If CSMCs cannot relax—due to calcium imbalance, oxidative stress, or apoptosis—blood cannot fill the sinusoids, and venous occlusion fails.
2. Why do PDE5 inhibitors sometimes fail?
PDE5 inhibitors require functional smooth muscle to exert their effect. In cases of fibrosis, severe endothelial dysfunction, or apoptosis-driven muscle loss, cGMP elevation cannot restore rigidity.
3. Can CSMC dysfunction be reversed?
Early dysfunction may be reversible through lifestyle changes, endothelial therapies, PDE5 inhibitors, and metabolic control. Advanced fibrosis is far more difficult to reverse and often requires regenerative treatments under investigation.
