Introduction: When Dust Leaves a Vascular Signature
Silicosis is often described in the language of fibrosis, nodules, and restrictive physiology. It is a disease of dust inhalation, progressive scarring, and long-term occupational exposure. Yet in some patients, the consequences extend beyond interstitial damage and into the pulmonary vasculature, where the clinical picture becomes more intricate—and more dangerous.
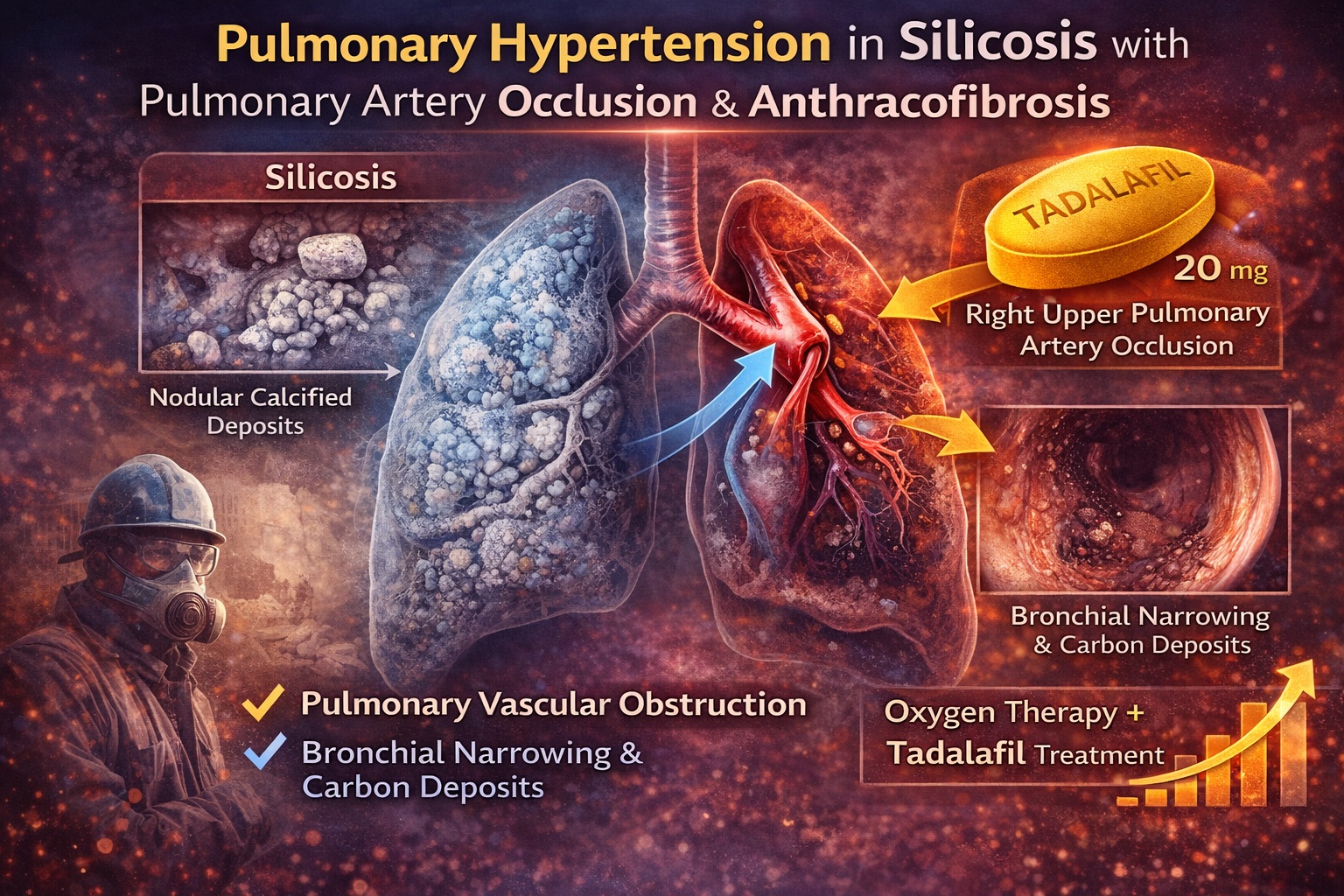
The case reported in Respiratory Medicine Case Reports describes an 81-year-old man with silicosis who developed pulmonary hypertension (PH) due to right upper pulmonary artery occlusion in association with bronchial anthracofibrosis . This combination is rare. Bronchial anthracofibrosis is defined by bronchial stenosis with black pigmentation of the mucosa, typically related to dust exposure or tuberculosis, and is not usually associated with arterial occlusion .
The convergence of silicosis, extrinsic pulmonary artery compression, bronchial narrowing, and pulmonary hypertension in a single patient presents a powerful illustration of how occupational lung disease can evolve beyond parenchymal scarring into vascular compromise. This article examines the pathophysiology, diagnostic approach, imaging findings, hemodynamic implications, and therapeutic considerations—including the use of tadalafil as a phosphodiesterase-5 inhibitor in PH due to lung disease.
Silicosis: From Nodules to Vascular Remodeling
Silicosis results from inhalation of crystalline silica particles, most commonly in mining, construction, or stone processing. The patient in the case had worked in a coal mine from age 20 to 30 and had long-standing silicosis diagnosed years earlier . Radiologically, his chest imaging showed nodular shadows, mediastinal and hilar lymphadenopathy with classic “egg-shell” calcification, and stable interstitial changes over time .
While fibrosis and lymph node calcification are hallmarks of silicosis, vascular involvement is less frequently emphasized. Experimental models suggest that silica exposure induces endothelial dysfunction, inflammation, and vascular remodeling, contributing to pulmonary hypertension . In addition to hypoxic vasoconstriction from parenchymal damage, structural vascular compromise may occur.
In this patient, contrast-enhanced CT demonstrated right upper pulmonary arterial stenosis and dilation of the main pulmonary artery. The pulmonary artery to ascending aorta ratio exceeded 1, a radiologic marker suggestive of PH . Importantly, no thrombus was identified.
Thus, the vascular burden in silicosis may arise not only from diffuse remodeling but also from localized compression.
Bronchial Anthracofibrosis: Black Pigment and Fibrotic Narrowing
Bronchial anthracofibrosis is defined by bronchial stenosis accompanied by black mucosal pigmentation . On bronchoscopy, the patient demonstrated carbon powder deposits and narrowing, predominantly in the right upper lobe bronchus (Figure 4 in the report) .
The mechanism is believed to involve peribronchial lymph node enlargement with extrinsic compression and fibrotic invasion into adjacent bronchi. Silica exposure is a recognized contributor. While tuberculosis has historically been associated, mineral dust exposure alone can produce this pattern.
The novelty in this case lies in the anatomical concordance: the bronchial anthracofibrosis occurred in the same lobe as the pulmonary artery occlusion. Coronal CT imaging demonstrated extrinsic compression of the right upper pulmonary artery .
This spatial overlap strongly suggests a shared mechanism—likely lymphadenopathy compressing both airway and artery.
Hemodynamic Confirmation: Defining Pulmonary Hypertension
Pulmonary hypertension is not diagnosed by imaging alone. Right heart catheterization (RHC) remains the gold standard.
In this patient, RHC demonstrated:
- Mean pulmonary arterial pressure (mPAP): 32 mmHg
- Pulmonary vascular resistance (PVR): 4.69 Wood units
- Pulmonary artery wedge pressure (PAWP): 12 mmHg
- Cardiac index: 2.62 L/min/m²
These findings confirm precapillary pulmonary hypertension, consistent with PH due to lung disease (Group 3 PH).
Ventilation-perfusion scanning further clarified the mechanism. Ventilation was preserved, but perfusion scintigraphy revealed a perfusion defect in the right upper lobe . Pulmonary angiography confirmed occlusion of the right upper pulmonary artery without thromboembolism.
The absence of thrombus excludes chronic thromboembolic PH. Instead, extrinsic compression and structural vascular compromise appear responsible.
Pathophysiological Integration: A Converging Mechanism
This case illustrates three interacting processes:
- Silicosis causing parenchymal and lymph node fibrosis
- Bronchial anthracofibrosis producing airway narrowing
- Extrinsic compression of the right upper pulmonary artery leading to localized perfusion loss
Silica exposure has been associated with endothelial injury and vascular remodeling in animal models . Enlarged calcified lymph nodes may compress adjacent vascular structures.
When compression reduces arterial lumen diameter, regional perfusion decreases. Chronic hypoperfusion may trigger compensatory vasoconstriction and elevate pulmonary vascular resistance.
The result is a form of PH that is not solely diffuse hypoxic vasoconstriction, but partially mechanical obstruction.
Clinical Manifestations: Subtle Onset, Significant Impact
The patient presented with exertional dyspnea and reduced six-minute walk distance (6MWD) of 246 meters, with oxygen desaturation to 88% during exertion . Echocardiography showed elevated tricuspid regurgitation pressure gradient (48 mmHg), prompting further investigation.
Laboratory markers revealed elevated NT-proBNP, indicating right ventricular strain . Such findings reflect hemodynamic burden rather than inflammatory progression.
Importantly, his CT findings had not significantly changed compared to previous imaging. This emphasizes that vascular deterioration may occur even when parenchymal radiology appears stable.
Clinicians managing silicosis must remain alert to evolving vascular complications despite static imaging.
Therapeutic Approach: Oxygen and PDE5 Inhibition
There is no curative therapy for silicosis or bronchial anthracofibrosis . Management focuses on symptom control and complication mitigation.
The patient was treated with:
- Home oxygen therapy during exertion
- Tadalafil 20 mg daily (a phosphodiesterase-5 inhibitor)
Oxygen therapy can attenuate hypoxic vasoconstriction. Phosphodiesterase-5 inhibitors such as tadalafil enhance nitric oxide–mediated vasodilation by increasing cyclic GMP levels in pulmonary vascular smooth muscle.
After six months, mPAP remained stable (32–33 mmHg), but 6MWD improved from 246 to 285 meters . Symptomatically, the patient improved.
This represents the first reported case of combined oxygen therapy and PDE5 inhibitor use in PH due to silicosis with pulmonary artery occlusion and bronchial anthracofibrosis .
Tadalafil in Group 3 Pulmonary Hypertension: A Delicate Balance
Pulmonary hypertension due to lung disease (Group 3 PH) is controversial territory for vasodilator therapy. Excess vasodilation may worsen ventilation-perfusion mismatch.
However, in selected patients with disproportionate vascular involvement, PDE5 inhibitors may improve functional capacity. Tadalafil, with its once-daily dosing and sustained pulmonary vasodilatory effect, offers practical advantages.
In this case, gas exchange did not deteriorate. Instead, functional improvement occurred without hemodynamic worsening.
This suggests that in carefully selected patients—particularly those with focal vascular obstruction—PDE5 inhibition may be beneficial.
Broader Implications: Anthracofibrosis as a High-Risk Marker
Studies cited in the report suggest that over 50% of patients with bronchial anthracofibrosis may have echocardiographic evidence of PH . While RHC confirmation was not universally performed in prior cohorts, this association is noteworthy.
Bronchial anthracofibrosis may therefore serve as a clinical marker of increased PH risk.
Given its association with lymph node enlargement and potential vascular compression, clinicians should consider screening for PH in patients with significant anthracofibrotic changes.
Early identification may allow timely intervention.
Conclusion: When Airway, Artery, and Dust Converge
This case highlights a rare but instructive scenario: pulmonary hypertension arising from silicosis compounded by localized pulmonary artery occlusion and bronchial anthracofibrosis .
The imaging correlation between airway stenosis and arterial compression underscores the importance of anatomical precision in diagnosis. Right heart catheterization confirmed precapillary PH. Oxygen therapy and tadalafil stabilized hemodynamics and improved functional capacity.
Silicosis is not merely a fibrotic disease. It can distort anatomy, compress vessels, and reshape pulmonary circulation.
For clinicians managing occupational lung disease, vigilance for vascular complications is essential. And in selected cases, targeted vasodilator therapy may offer meaningful benefit.
FAQ
1. How does silicosis cause pulmonary hypertension?
Silicosis can lead to PH through hypoxic vasoconstriction from lung fibrosis, endothelial injury, vascular remodeling, and—in rare cases—extrinsic compression of pulmonary arteries by enlarged lymph nodes.
2. What is bronchial anthracofibrosis?
It is bronchial narrowing with black mucosal pigmentation, typically due to dust exposure or tuberculosis. It may reflect lymph node–related compression and can be associated with increased PH risk.
3. Can tadalafil be used in pulmonary hypertension due to lung disease?
While controversial, PDE5 inhibitors such as tadalafil may improve exercise capacity in selected patients with Group 3 PH. Careful monitoring is essential to avoid worsening gas exchange.