

Introduction: When Familiar Drugs Reveal Unexpected Biology
Phosphodiesterase type 5 inhibitors (PDE5Is) such as sildenafil, vardenafil, and tadalafil have become synonymous with the treatment of erectile dysfunction. Their mechanism is well understood, their clinical use widespread, and their safety profile—at least at the surface—reassuring.
Yet pharmacology rarely confines itself to a single organ or function. Drugs designed to act in one tissue often leave biochemical footprints elsewhere. The liver, as the central hub of drug metabolism, is particularly sensitive to such influences.
The study offers an important shift in perspective. It examines how erectile dysfunction drugs alter hepatic enzyme systems, specifically cytochrome P450 (CYP) enzymes and related metabolic pathways in male rats. What emerges is not merely a pharmacokinetic curiosity, but a deeper understanding of how these widely used agents may influence drug metabolism, toxicity, and systemic physiology.
This article expands on those findings, translating experimental insights into clinically meaningful implications—with particular attention to tadalafil, a drug often considered pharmacologically elegant, yet not entirely innocent.
Cytochrome P450: The Silent Gatekeeper of Drug Metabolism
The cytochrome P450 enzyme system is one of the most sophisticated biochemical networks in the human body. It governs the metabolism of endogenous compounds—such as hormones and fatty acids—as well as exogenous substances, including medications, toxins, and carcinogens.
These enzymes are not static. Their expression and activity fluctuate in response to environmental factors, diet, disease states, and, importantly, other drugs.
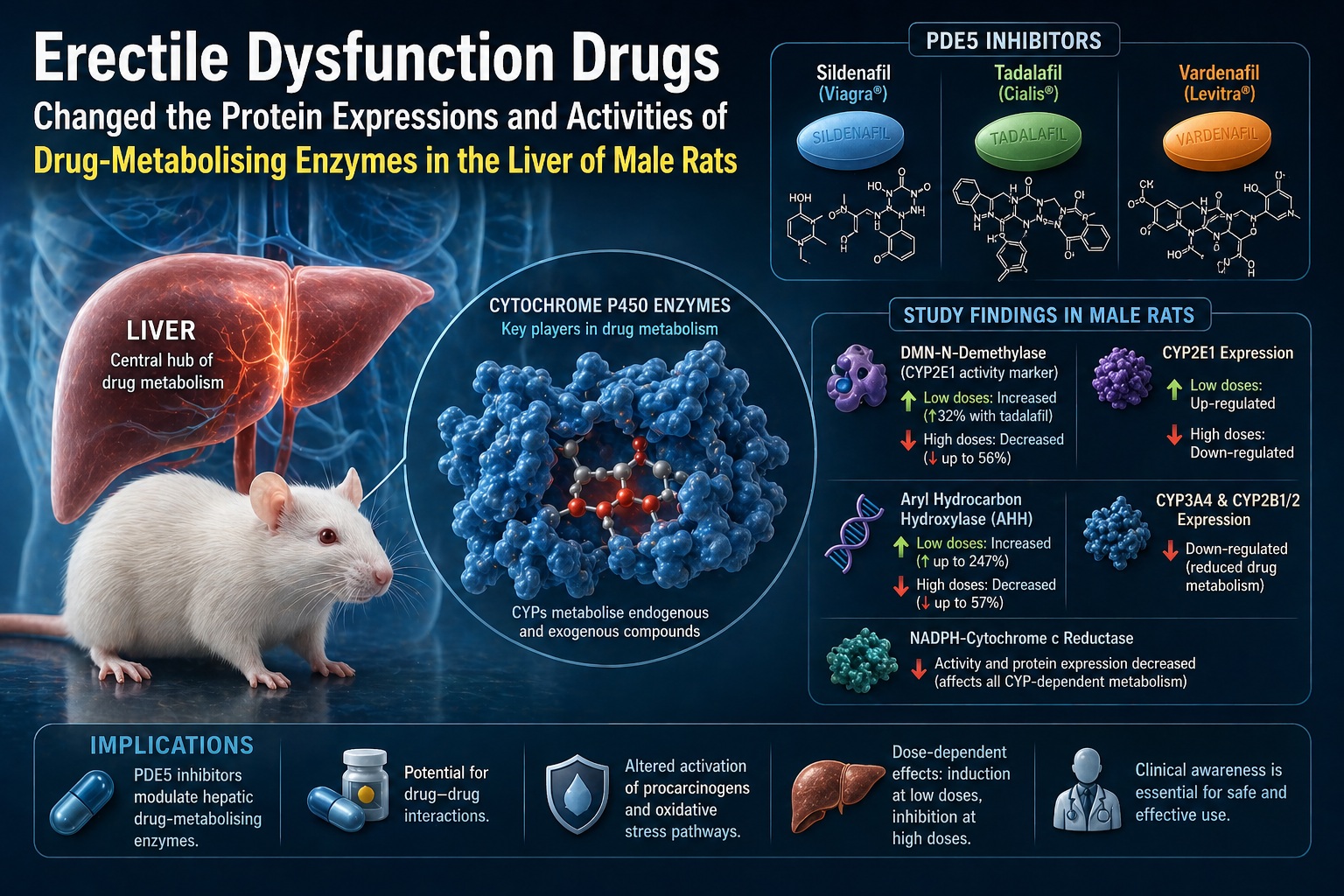
As demonstrated in the study, PDE5 inhibitors significantly alter both the expression and activity of multiple CYP isoforms, including CYP1A2, CYP2E1, CYP3A4, and CYP2C subfamilies .
This matters because CYP enzymes do not operate in isolation. A change in one pathway may amplify or suppress another, creating a cascade of metabolic consequences. In practical terms, this means that a drug taken for erectile dysfunction may subtly influence how the body processes entirely unrelated medications.
The liver, in this context, becomes less of a passive processor and more of a dynamic participant in pharmacological interactions.
Dose Matters: The Paradox of Induction and Inhibition
One of the most intriguing findings of the study is the dose-dependent duality of PDE5 inhibitors.
At low doses, tadalafil and related drugs tend to induce certain enzyme activities. For example, low-dose tadalafil increased dimethylnitrosamine N-demethylase (DMN-dI) activity by approximately 32% . This suggests enhanced metabolic activation of certain compounds.
At higher doses, however, the effect reverses. The same drugs inhibit enzyme activity—sometimes dramatically. High-dose tadalafil reduced DMN-dI activity by nearly 50%, while also suppressing key CYP isoforms.
This biphasic response is not merely academic. It reflects the complex binding behavior of CYP enzymes, which may accommodate multiple substrate molecules simultaneously. At higher concentrations, drugs may occupy inhibitory binding sites, reducing enzymatic efficiency.
In simpler terms: a little may stimulate metabolism; too much may suppress it.
This paradox introduces an important clinical nuance—dose is not just about efficacy, but about metabolic consequences.
Tadalafil and CYP2E1: A Double-Edged Relationship
CYP2E1 is a particularly significant enzyme, involved in the metabolism of small organic molecules, including ethanol and various carcinogens.
The study demonstrates that low doses of tadalafil increase CYP2E1 expression, while high doses suppress it .
This finding carries two opposing implications:
- Induction of CYP2E1 may enhance the generation of reactive oxygen species (ROS), increasing oxidative stress and potential cellular damage.
- Inhibition of CYP2E1, conversely, may reduce the activation of carcinogenic compounds and protect tissues from toxicity.
Thus, tadalafil operates in a delicate balance—potentially promoting oxidative pathways at one dose and suppressing them at another.
From a clinical perspective, this reinforces the importance of appropriate dosing and highlights the potential for long-term metabolic effects that extend beyond erectile function.
Impact on Carcinogen Metabolism: A Subtle but Significant Risk
The liver’s role in carcinogen metabolism is mediated largely by CYP enzymes such as CYP1A1, CYP1A2, and CYP2E1.
The study reveals that PDE5 inhibitors significantly alter aryl hydrocarbon hydroxylase (AHH) activity, a key enzyme involved in the activation of polycyclic aromatic hydrocarbons (PAHs)—known carcinogens.
At low doses, certain PDE5 inhibitors increased AHH activity dramatically—by up to 247% in the case of sildenafil . At higher doses, the same activity was suppressed.
This creates a complex biological scenario. Increased AHH activity may enhance the activation of carcinogens, while decreased activity may reduce it but simultaneously impair detoxification pathways.
Tadalafil, within this framework, behaves as a modulator rather than a simple inhibitor or inducer.
The implication is not that these drugs cause cancer—far from it—but that they may influence the body’s handling of carcinogenic exposures in subtle ways.
CYP3A4 and Drug Interactions: The Clinical Core
Among all CYP enzymes, CYP3A4 holds particular clinical importance. It is responsible for metabolizing a vast array of medications, from antibiotics to statins and immunosuppressants.
The study shows that tadalafil, along with other PDE5 inhibitors, inhibits CYP3A4 expression .
This has direct clinical consequences. Reduced CYP3A4 activity can lead to:
- Increased plasma levels of co-administered drugs
- Prolonged drug half-life
- Enhanced risk of adverse effects
Conversely, drugs that induce CYP3A4 may reduce tadalafil’s effectiveness by accelerating its clearance.
This interplay underscores the importance of medication review in patients using PDE5 inhibitors. What appears to be a simple prescription may, in reality, be part of a complex pharmacological network.
Reductase Suppression: A Hidden Layer of Metabolic Control
Beyond CYP enzymes themselves, the study highlights the suppression of NADPH-cytochrome c reductase, a key enzyme that supplies electrons to the CYP system.
Both low and high doses of tadalafil significantly reduced this enzyme’s activity .
This finding is particularly important because it affects the entire CYP system, not just individual enzymes. Without adequate reductase activity, CYP-mediated metabolism becomes inefficient.
The result is a broad alteration in metabolic capacity—affecting not only drugs but also endogenous processes such as steroid synthesis and lipid metabolism.
In other words, tadalafil may influence not just specific pathways, but the overall metabolic tone of the liver.
Clinical Translation: What Do These Findings Mean for Patients?
Translating animal data into clinical practice requires caution, but the implications are nonetheless compelling.
For patients, the key message is not alarm, but awareness. PDE5 inhibitors are effective and generally safe, but they are not metabolically inert.
Clinicians should consider:
- Potential drug-drug interactions, especially with CYP3A4 substrates
- The impact of dose on metabolic pathways
- The possibility of altered responses to other medications
For long-term users of tadalafil, particularly those with comorbidities requiring multiple medications, this becomes especially relevant.
The liver, after all, does not compartmentalize its responsibilities.
A Broader Perspective: Rethinking “Simple” Drugs
It is tempting to categorize drugs as simple or complex, based on their clinical use. PDE5 inhibitors, given their targeted indication, often fall into the former category.
This study challenges that assumption.
By demonstrating widespread effects on hepatic enzyme systems, it reveals a level of biochemical complexity that extends far beyond erectile function.
Tadalafil, in particular, emerges as a drug with systemic influence—capable of modulating oxidative stress, enzyme expression, and metabolic pathways.
This does not diminish its value. Rather, it enriches our understanding and encourages more thoughtful use.
Conclusion: Precision in Prescription, Awareness in Practice
The interaction between PDE5 inhibitors and liver metabolism represents a subtle but important dimension of modern pharmacology.
Tadalafil and its counterparts are not merely facilitators of vascular relaxation—they are modulators of biochemical systems that influence drug metabolism, oxidative balance, and potentially long-term health outcomes.
The study reminds us that even well-established drugs deserve continued scrutiny.
In clinical practice, this translates into a simple principle: prescribe with precision, monitor with awareness, and respect the complexity of the human body.
Because in medicine, simplicity is often an illusion—and understanding is always evolving.
FAQ: Key Questions About PDE5 Inhibitors and Liver Metabolism
1. Do PDE5 inhibitors affect liver enzymes?
Yes, they can alter the expression and activity of cytochrome P450 enzymes, especially CYP3A4 and CYP2E1.
2. Is tadalafil safe for long-term use?
Generally yes, but long-term use should consider potential interactions with other medications metabolized in the liver.
3. Can these drugs increase toxicity of other medications?
Potentially, especially if they inhibit enzymes responsible for drug clearance.
4. Are these effects dose-dependent?
Yes, low and high doses can have opposite effects on enzyme activity.
5. Should patients be concerned?
Not necessarily—but clinicians should be aware and manage therapy accordingly.
