Cyclic nucleotides—cAMP and cGMP—have long been celebrated as the currency of intracellular signaling, orchestrating processes as diverse as vasodilation, neurotransmission, gene regulation, immune activation, and apoptosis. Their synthesis and degradation have been thoroughly dissected, with phosphodiesterases (PDEs) cemented as central regulators. Yet, as the work underlying this review makes abundantly clear, intracellular concentration is only part of the story. Cyclic nucleotides are also actively transported across membranes by a pair of high-affinity, ATP-dependent efflux pumps: ABCC4 (MRP4) and ABCC5 (MRP5).
The paper’s authors revisit a long-overlooked question: What happens when PDE inhibitors—both specific and non-specific—interact with these transporters? Given that many PDE inhibitors are frequently administered in clinical practice, and given that transporter-mediated efflux contributes critically to cyclic nucleotide compartmentalization, this is not an esoteric inquiry but a pharmacological necessity.
This article synthesizes the paper’s key findings, providing a broader scientific context and a translational interpretation. The interplay between PDE inhibition and transporter modulation reveals a subtle, layered regulatory system—one in which drugs designed to elevate intracellular cAMP or cGMP may simultaneously influence (or be influenced by) the transporters that dictate their distribution. The implications ripple outward: toward cardiovascular physiology, oncology, inflammation, neuropharmacology, and beyond.
Cyclic Nucleotide Transporters: The Often-Ignored Gatekeepers of Second Messenger Dynamics
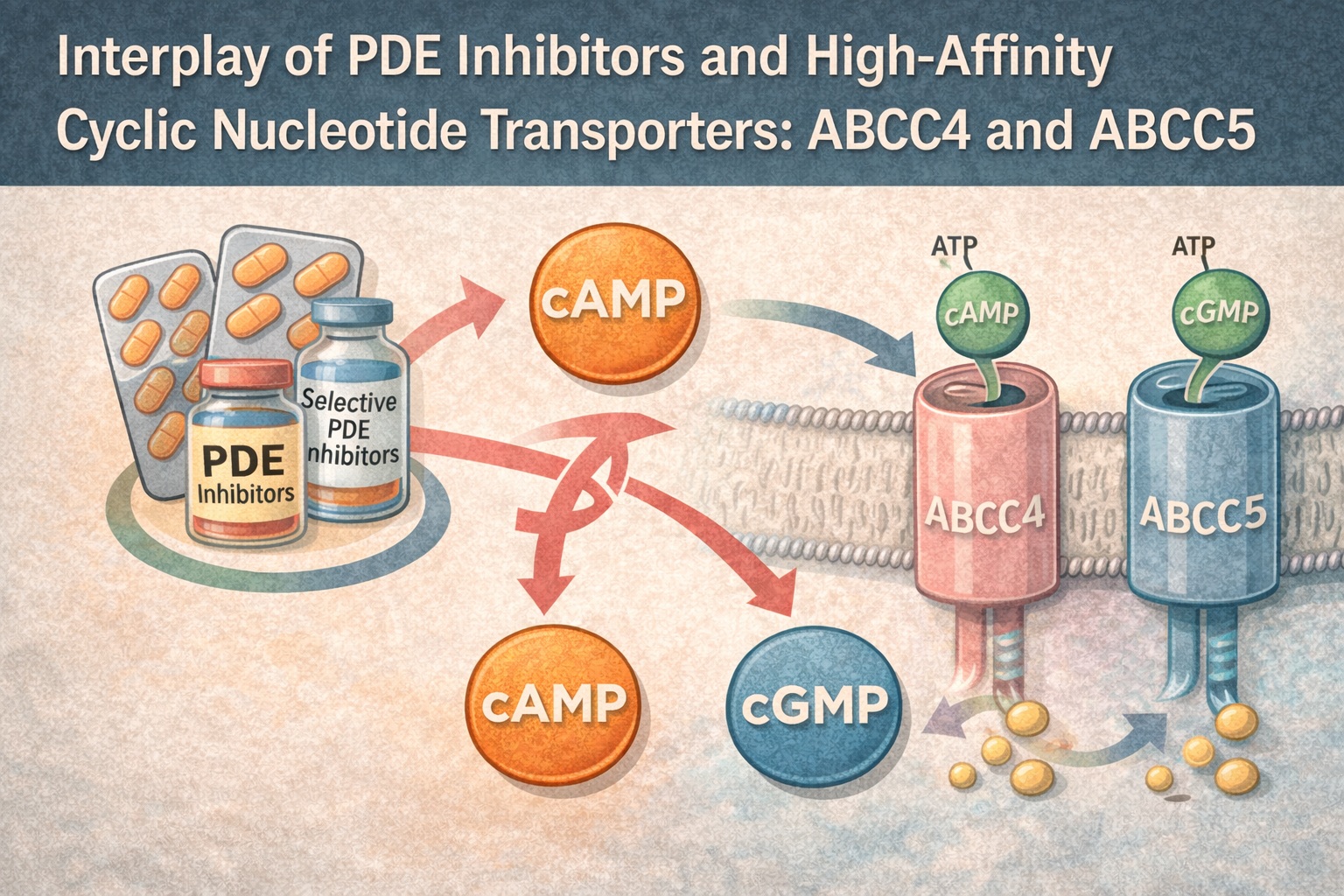
Intracellular cyclic nucleotide signaling is dynamic, spatially segregated, and highly regulated. PDEs are traditionally viewed as the primary means of deactivating cAMP and cGMP, yet ABCC4 and ABCC5 contribute significantly to the spatial compartmentalization of these messengers by actively exporting them out of cells.
ABCC4 and ABCC5 are members of the ATP-Binding Cassette (ABC) transporter family, utilizing ATP hydrolysis to pump cyclic nucleotides across plasma and intracellular membranes. The authors highlight that ABCC4 preferentially transports cAMP, while ABCC5 has a higher affinity for cGMP, although both transport both substrates to some degree. In vitro assays in membrane vesicles (page 3, Fig. 1) demonstrate clear differences in Km values, providing biochemical validation of their substrate specialization.
This transporter activity is not simply a passive clearance mechanism. Efflux regulates localized cyclic nucleotide pools near membrane microdomains, fine-tuning activation of protein kinase A (PKA), protein kinase G (PKG), and Epac. Transporter dysfunction or inhibition therefore reshapes intracellular signaling topography.
The authors emphasize that cyclic nucleotide efflux also generates extracellular signaling loops—a point often ignored in standard pharmacology. Extracellular cAMP and cGMP can act as paracrine modulators or be degraded into adenosine, which activates adenosine receptors with potent anti-inflammatory and vasodilatory effects. Thus, transporter activity shapes not only intracellular but intercellular communication.
The paper stands out because it directly examines how PDE inhibitors, intended to elevate cyclic nucleotide levels, interact with and modulate ABCC4 and ABCC5. This is a fundamental yet neglected aspect of pharmacology: PDE inhibitors do not act on a static system but on a dynamic, transporter-buffered environment whose response can deviate from textbook predictions.
How PDE Inhibitors Interact with Cyclic Nucleotide Transporters: Mechanistic Insights
One of the study’s central findings is that PDE inhibitors themselves influence ABCC4 and ABCC5—either by acting as transported substrates, partial inhibitors, or competitive binders. Using ATP-dependent transport assays, the authors quantify how different PDE inhibitors alter cyclic nucleotide efflux, revealing patterns that are as intriguing as they are clinically relevant.
The first major insight is that many PDE inhibitors inhibit cyclic nucleotide transport at concentrations overlapping with their PDE-inhibitory ranges. For example, non-selective agents like IBMX inhibit both ABCC4 and ABCC5, reducing efflux in a dose-dependent manner. This means that intracellular cyclic nucleotide accumulation under IBMX may result not only from reduced degradation but also reduced export—an important distinction for interpreting cAMP/cGMP–dependent responses.
Additionally, the study explores whether PDE inhibitors themselves are transported by ABCC4 or ABCC5. While some compounds did not appear to be transported (as shown on page 5, substrate competition assays), others appeared to act as competitive blockers, binding transporter sites without being efficiently moved across the membrane. This competitive inhibition suggests that PDE inhibitors may alter cyclic nucleotide availability not merely by elevating production or slowing degradation but by bottlenecking the transporter-mediated escape route.
The mechanistic nuance is meaningful. When a PDE inhibitor blocks efflux, intracellular cAMP or cGMP may rise more sharply and persist longer, potentially leading to stronger or more prolonged PKA or PKG activation than predicted based solely on PDE activity. This has implications for vascular tone, immune cell activation, platelet aggregation, and neuronal excitability—systems exquisitely sensitive to cyclic nucleotide flux.
Ultimately, the study reframes the pharmacodynamic landscape of PDE inhibitors: they do not act solely by inhibiting PDEs but by reshaping the transporter environment controlling second messenger compartmentalization.
Selective PDE Inhibitors: Differential Modulation of ABCC4 and ABCC5
A key strength of the paper lies in its systematic evaluation of selective PDE inhibitors. By testing inhibitors targeting PDE3, PDE4, and PDE5, the researchers provide clarity on which subtypes modulate which transporters, and to what extent.
PDE4 inhibitors (e.g., rolipram) exhibited mild modulation of ABCC4-mediated cAMP transport, but minimal impact on ABCC5. The data suggest that PDE4 inhibitors may potentiate local cAMP signaling by reducing degradation more effectively than export, creating a strongly compartmentalized cAMP pool.
PDE5 inhibitors (tadalafil, sildenafil, vardenafil) showed a more complex profile. While they potently inhibit PDE5 and raise intracellular cGMP, their impact on ABCC5 was minimal compared to non-selective inhibitors. Interestingly, some PDE5 inhibitors showed moderate interference with ABCC4’s cAMP transport. This implies that PDE5 inhibitors might indirectly modulate cAMP-dependent pathways—not through PDE3 or PDE4, but through transporter crosstalk. Clinically, this may partially explain why PDE5 inhibitors demonstrate anti-inflammatory, anti-fibrotic, and metabolic benefits independent of cGMP alone.
PDE3 inhibitors had the broadest transporter interaction footprint, altering both ABCC4 and ABCC5 more significantly than PDE4 or PDE5 inhibitors. Because PDE3 hydrolyzes both cyclic nucleotides, its inhibitors naturally create biochemical ripple effects—but the transporter modulation shown in the study suggests that PDE3 blockade alters efflux at least as much as intracellular hydrolysis.
These findings highlight an underappreciated principle: the pharmacological purity of selective PDE inhibitors is compromised at the level of transporter modulation. Drugs marketed as “selective” based on enzyme assays may behave less selectively in live cellular environments shaped by active transport processes.
Non-Specific PDE Inhibitors: Broad-Range Modulators with Wide Physiological Reach
The non-specific PDE inhibitors examined in the study—including IBMX and theophylline—demonstrated the strongest transporter inhibition across both ABCC4 and ABCC5. Their capacity to elevate cyclic nucleotide levels arises from:
- inhibition of multiple PDE families,
- inhibition of cyclic nucleotide efflux,
- competitive binding to transporters that reshapes intracellular signaling kinetics.
This combined mechanism explains why drugs like theophylline have wide, sometimes unpredictable systemic effects. Their transporter interaction amplifies intracellular signaling in ways that enzyme inhibition alone cannot predict.
The authors note that at concentrations often achieved therapeutically or sub-therapeutically, non-specific PDE inhibitors can significantly reduce cyclic nucleotide transport (page 6, concentration–response curves). This finding provides insight into the notorious narrow therapeutic window of theophylline, where small changes in drug exposure produce large and sometimes dangerous shifts in intracellular signaling.
From a translational perspective, these data underscore why historically “dirty” PDE inhibitors have such wide-ranging physiological influence: they manipulate both sides of cyclic nucleotide homeostasis—degradation and efflux. This dual influence amplifies therapeutic potency but increases risk when not carefully controlled.
Broader Biological and Translational Implications: Cyclic Nucleotide Transport as a Pharmacological Multiplier
The final section of the article elegantly ties together the implications of transporter modulation. When PDE inhibitors also inhibit cyclic nucleotide transporters, a synergistic amplification of signaling occurs. This amplification may be beneficial, neutral, or harmful depending on tissue type and clinical context.
In vascular smooth muscle, elevated cGMP due to combined PDE5 inhibition and reduced efflux enhances vasodilation and may strengthen anti-proliferative signaling. This could be advantageous in pulmonary hypertension, systemic sclerosis, or early vascular remodeling.
In immune cells, ABCC4-mediated export of cAMP normally suppresses intracellular anti-inflammatory signaling. PDE inhibitors that block ABCC4 may therefore heighten cAMP-PKA–driven immunomodulation, potentially beneficial in chronic inflammation but problematic in infection.
In platelets, where cAMP and cGMP tightly regulate aggregation, transporter inhibition may reinforce antithrombotic effects—but also increase bleeding risk.
In neurons, exaggerated cAMP signaling may influence synaptic plasticity, possibly explaining both therapeutic cognitive benefits and adverse excitatory effects of certain PDE inhibitors.
These complex interactions emphasize the need for pharmacologists and clinicians to consider cyclic nucleotide homeostasis not as a single pathway but as a multidimensional system shaped by synthesis, degradation, transport, and compartmentalization. PDE inhibitors do not merely “increase cAMP” or “increase cGMP”—they reshape an entire network.
Conclusion
The study outlined in the provided article makes one central point unmistakably clear: PDE inhibitors regulate cyclic nucleotide signaling not only by modulating enzymatic degradation but also by modulating ATP-dependent efflux pathways. The high-affinity transporters ABCC4 and ABCC5 emerge as key players shaping the intracellular fate of cAMP and cGMP.
This reframing has wide-reaching consequences across cellular physiology and drug development. It implies that PDE inhibitor pharmacodynamics involve transporter interactions overlooked in traditional analyses. It explains clinical observations that could not be understood by PDE inhibition alone. And it opens new questions about optimizing cyclic nucleotide–targeting therapies across inflammation, fibrosis, oncology, metabolism, and cardiovascular disease.
As the authors argue, understanding transporter modulation is not optional—it is required for accurate prediction of drug behavior. For a pharmacological field long dominated by enzyme-centric thinking, this represents a welcome and necessary evolution.
FAQ
1. Do PDE inhibitors raise cyclic nucleotide levels partly by blocking their transport?
Yes. This study shows that many PDE inhibitors—especially non-selective ones—directly inhibit ABCC4 and ABCC5 efflux, amplifying intracellular cAMP and cGMP accumulation beyond the effect of PDE inhibition alone.
2. Are selective PDE inhibitors (e.g., PDE5 inhibitors) truly selective at the transporter level?
Not entirely. Even selective inhibitors modulate transporters to some extent. For example, PDE5 inhibitors may interfere with ABCC4-mediated cAMP transport despite being selective for PDE5 enzymatically.
3. Why is transporter modulation clinically relevant?
Because efflux determines spatial and temporal dynamics of cyclic nucleotide signaling. Transporter modulation influences vascular reactivity, immune cell behavior, platelet aggregation, neuronal signaling, and responses to inflammatory stress.