
Introduction: Beyond Classical PDE5 Inhibition
Few small molecules have reshaped modern pharmacotherapy as profoundly as tadalafil. Originally developed as a highly selective phosphodiesterase type 5 (PDE5) inhibitor for erectile dysfunction, tadalafil rapidly expanded its clinical relevance to pulmonary arterial hypertension and lower urinary tract symptoms associated with benign prostatic hyperplasia. Its long half-life, high selectivity, and favorable safety profile have made it a cornerstone molecule in vascular medicine.
Yet pharmacology rarely stands still. Over the past decade, tadalafil has evolved from a therapeutic endpoint into a structural template. The rationale is compelling: if a rigid, highly optimized PDE5 inhibitor demonstrates such diverse biological properties—including emerging anticancer potential—then carefully designed structural modifications may uncover entirely new pharmacodynamic profiles.
The study analyzed in this article explored precisely that strategy. By replacing the rigid pyrazinopyridoindole core of tadalafil with a more flexible diketopiperazine scaffold, researchers sought to understand how conformational flexibility, stereochemistry, and halogen substitution influence biological activity. The results challenge assumptions about structure–activity relationships and open new avenues in medicinal chemistry.
This article examines the chemical design, crystallographic insights, enzymatic findings, cytotoxic effects, and broader therapeutic implications of these novel tadalafil analogues.
Rationale for Structural Modification of Tadalafil
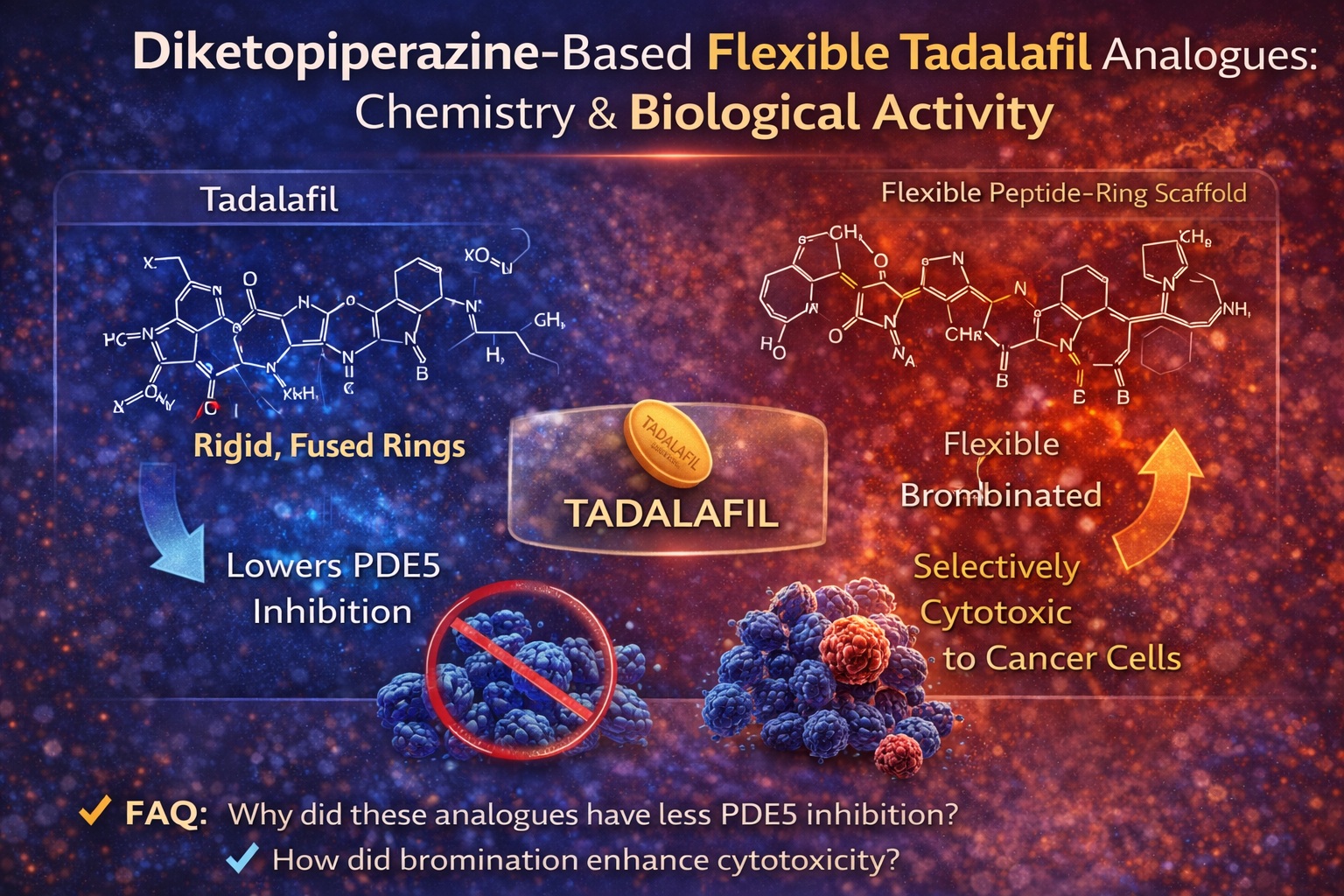
Tadalafil’s pharmacological potency arises from its precise three-dimensional fit within the catalytic pocket of PDE5. The rigid fused-ring system locks the molecule into a conformation optimal for binding. While this rigidity enhances specificity, it may also restrict adaptability toward alternative biological targets.
The investigators pursued three principal modifications:
- Introduction of a flexible diketopiperazine scaffold
- Variation in stereochemistry via D- and L-tryptophan derivatives
- Bromine substitution within the aromatic moiety
The substitution of the rigid heterocyclic core with a diketopiperazine ring system represents a fundamental architectural shift. Diketopiperazines are recognized “privileged structures” in medicinal chemistry. They appear in numerous bioactive compounds and frequently serve as scaffolds capable of engaging diverse protein targets.
By increasing molecular flexibility, the researchers aimed to test whether dynamic conformations could enhance binding to alternative biological targets—particularly those involved in tumor biology—while possibly preserving or modulating PDE5 interaction.
The approach reflects a deeper medicinal chemistry principle: rigidity often enhances specificity, whereas flexibility may enhance adaptability. Determining where therapeutic advantage lies requires experimental evidence rather than theoretical speculation.
Synthetic Strategy and Chemical Design
The synthetic route was carefully constructed, beginning with protected tryptophan derivatives. Dipeptide formation, cyclization to the diketopiperazine intermediate, N-benzylation, and final deprotection yielded four final analogues:
- 6a and 6b (non-brominated enantiomers)
- 7a and 7b (brominated enantiomers)
The use of cesium carbonate proved essential for successful N-alkylation, highlighting the importance of base selection in optimizing reaction yields. Removal of protective groups via trifluoroacetic acid completed the synthesis.
From a structural standpoint, these analogues differ from tadalafil in several critical ways:
- The fused rigid core is replaced with a disjointed, conformationally flexible diketopiperazine system
- Indole and dioxole moieties are no longer locked into a single spatial orientation
- Brominated derivatives introduce potential halogen-bonding interactions
These modifications are not superficial. They alter torsional angles, intermolecular interactions, and potentially receptor-binding dynamics.
The elegance of the synthetic work lies not merely in chemical execution, but in the deliberate design of structural variables that can be correlated with biological behavior.
Crystallographic Insights: Rigidity Versus Flexibility
Single-crystal X-ray diffraction studies provided crucial structural information. Unlike tadalafil’s compact, rigid architecture, the newly synthesized analogues exhibited noticeable conformational freedom between the indole and diketopiperazine fragments.
Crystallographic analysis demonstrated:
- Variable inclination angles between ring systems
- Distinct intermolecular hydrogen bonding networks
- Weak C–H···O and C–H···π interactions stabilizing layered supramolecular architectures
In tadalafil, the fused core maintains a fixed spatial relationship between functional groups. In contrast, compounds 6a, 6b, 7a, and 7b display bending of the diketopiperazine moiety relative to the indole ring, suggesting increased flexibility in solution as well.
From a pharmacological perspective, this structural flexibility may reduce the precision of PDE5 binding while potentially allowing adaptation to other protein targets.
The crystallographic superposition of tadalafil with its analogues vividly illustrates this difference: where tadalafil is architecturally disciplined, the new analogues are structurally exploratory.
PDE5 Inhibition: A Clear Loss of Potency
Despite being designed from a potent PDE5 inhibitor, the new compounds demonstrated markedly reduced activity against human PDE5.
Tadalafil displayed nanomolar affinity, consistent with established literature. In contrast, the most potent analogue (7a) exhibited inhibitory constants more than 300-fold weaker than tadalafil.
Several conclusions emerge:
- Conformational rigidity is critical for optimal PDE5 inhibition
- Flexibility disrupts precise alignment within the catalytic pocket
- Bromination modestly enhances inhibition compared to non-brominated analogues
- Stereochemistry influences activity, though not dramatically
The data confirm that tadalafil’s rigid pyrazinopyridoindole core is not merely structural ornamentation—it is central to enzymatic potency.
Interestingly, brominated derivatives (7a, 7b) consistently outperformed their non-halogenated counterparts. This suggests that halogen bonding or enhanced hydrophobic interactions may partially compensate for structural flexibility.
Nevertheless, none of the analogues approach tadalafil’s inhibitory strength.
Antiplatelet Activity: Functional Confirmation of Reduced PDE5 Inhibition
To evaluate functional consequences of PDE5 inhibition, the investigators assessed antiplatelet activity in the presence of nitric oxide.
Tadalafil significantly enhanced nitric oxide–mediated inhibition of platelet aggregation. The new analogues, however, failed to produce comparable effects.
This functional assay corroborates the enzymatic findings: the analogues do not meaningfully inhibit PDE5 in a physiological context.
The consistency between biochemical assay and platelet aggregation data strengthens the conclusion that structural flexibility compromises PDE5-targeted activity.
Cytotoxicity Across Cancer Cell Lines: An Unexpected Signal
If the story ended with loss of PDE5 inhibition, the analogues would be pharmacological disappointments. However, cytotoxicity testing across sixteen human cell lines revealed a more intriguing pattern.
Most cancer cell lines demonstrated limited sensitivity. Yet two cell types stood out:
- HEK 293T
- MCF7 (breast adenocarcinoma)
In these cells, compounds 7a and 7b exhibited greater cytotoxicity than tadalafil itself. The brominated analogues demonstrated the most consistent activity, with IC50 values approaching those of cisplatin in selected contexts—though still less potent overall.
Importantly:
- Cytotoxicity was selective rather than universal
- Bromine substitution correlated with enhanced activity
- The mechanism of cytotoxicity remains undefined
These findings suggest that the structural modifications, while detrimental for PDE5 inhibition, may have generated new biological interactions.
This shift—from vascular pharmacology to potential oncologic application—was not the primary objective, yet it may represent the most valuable outcome.
Toxicological Assessment in Caenorhabditis elegans
In vivo toxicity was assessed using Caenorhabditis elegans. At concentrations up to 160 µM, none of the compounds disrupted the nematode life cycle.
This finding suggests:
- Absence of acute organismal toxicity at tested doses
- A potentially favorable therapeutic window for further investigation
While C. elegans is not a substitute for mammalian toxicity testing, it provides an early indication that cytotoxic effects observed in tumor cells may not translate into nonspecific systemic toxicity.
Structure–Activity Relationships: Lessons from the Data
The study highlights several instructive structure–activity relationships:
- Rigidity strongly favors PDE5 inhibition
- Flexibility reduces enzymatic specificity
- Bromine substitution enhances cytotoxic potential
- Stereochemical configuration influences biological behavior
The paradox is instructive. What improves anticancer selectivity may diminish original enzymatic targeting.
In medicinal chemistry, this is not failure—it is evolution. A molecule designed for one target may become a scaffold for another.
Therapeutic Implications and Future Directions
The most promising candidates emerging from this work are compounds 7a and 7b. Their enhanced cytotoxic selectivity toward MCF7 cells, combined with low organismal toxicity in C. elegans, makes them reasonable starting points for anticancer lead optimization.
Future studies should focus on:
- Mechanistic elucidation of cytotoxic pathways
- Evaluation of apoptosis induction and cell cycle effects
- Target identification via proteomic approaches
- Optimization of halogen substitution patterns
- Exploration of additional heterocyclic modifications
It is particularly notable that tadalafil itself has been explored as an adjunct in cancer therapy, including immune modulation and enhancement of chemotherapeutic sensitivity. The present analogues may expand that domain beyond PDE5 inhibition.
In this sense, the work represents a pivot—from vascular pharmacology toward oncologic medicinal chemistry.
Broader Perspective: When Losing One Target Gains Another
Drug discovery often celebrates optimization toward a single target. Yet this study reminds us that structural diversification can reveal unexpected biological potential.
By relaxing the rigid core of tadalafil and introducing diketopiperazine flexibility, researchers lost PDE5 potency but gained selective cytotoxic behavior. This trade-off may prove valuable.
The medicinal chemist’s task is not only to preserve activity—but to explore structural space strategically. Sometimes, deviation from the original pharmacophore opens new therapeutic landscapes.
In that regard, these flexible tadalafil analogues represent not a failure of replication, but a proof of concept in scaffold evolution.
FAQ
1. Why did the new analogues lose PDE5 inhibitory potency compared to tadalafil?
Tadalafil’s high potency depends on its rigid fused-ring structure, which fits precisely into the PDE5 catalytic site. Replacing this rigid core with a flexible diketopiperazine scaffold altered spatial orientation and reduced optimal binding alignment, leading to markedly weaker inhibition.
2. Why are the brominated compounds (7a, 7b) more cytotoxic than the non-brominated ones?
Bromine can enhance hydrophobic interactions and form halogen bonds within protein binding pockets. These interactions may improve binding to alternative cellular targets involved in cancer cell survival, thereby increasing cytotoxic activity.
3. Could these analogues become anticancer drugs?
Potentially, yes—but substantial work remains. Compounds 7a and 7b demonstrated selective cytotoxicity in certain cancer cell lines and showed low toxicity in C. elegans. However, their mechanism of action is not yet defined, and further preclinical studies are required before considering clinical development.