Introduction
Among the many molecules that orchestrate life’s intricate biological symphony, nitric oxide (NO) is a paradoxical player. It is the smallest known signaling molecule, gaseous in nature, with a fleeting half-life measured in seconds. And yet, its influence stretches across the vascular, neural, and immune systems. Unlike hormones that rely on distant travel through the bloodstream, nitric oxide acts locally and almost instantly, diffusing across membranes to alter cell function. It dilates vessels, modulates neurotransmission, shapes immune defense, and—when dysregulated—can contribute to shock, neurodegeneration, and cardiovascular disease.
At the heart of this molecular performance are three enzymes: neuronal nitric oxide synthase (nNOS, NOS I), inducible nitric oxide synthase (iNOS, NOS II), and endothelial nitric oxide synthase (eNOS, NOS III). Each isoform works on the same fundamental chemical principle: converting the amino acid L-arginine into nitric oxide and L-citrulline, using oxygen and a suite of cofactors. Yet, their regulation, distribution, and outcomes differ profoundly, which makes them simultaneously indispensable for health and dangerous when unrestrained.
This article dives into the biology of nitric oxide synthases, unpacks their physiological functions, and explores how their dysfunction shapes human disease. We will also reflect on how modern pharmacology leverages their pathways—for better or worse.
The Chemistry Behind Nitric Oxide
To produce NO, each NOS enzyme operates as a dimer. Electrons flow from NADPH, through flavins FAD and FMN, to a heme group where oxygen is activated and used to oxidize L-arginine. The cofactor tetrahydrobiopterin (BH4) ensures that this reaction proceeds efficiently and without error. The result is the generation of NO and L-citrulline.
However, when cofactors are depleted or oxidative stress takes center stage, NOS can become “uncoupled.” Instead of producing NO, the enzyme leaks superoxide (O₂⁻), a reactive species that reacts with NO to form peroxynitrite (ONOO⁻), a potent oxidant that damages proteins, lipids, and DNA. Thus, the same enzyme that sustains vascular health may, under stress, accelerate vascular injury.
Regulation of NOS activity is also highly context-dependent. Neuronal and endothelial isoforms are calcium-dependent, activated when calmodulin binds in response to increased intracellular Ca²⁺. By contrast, inducible NOS binds calmodulin tightly regardless of calcium, ensuring continuous production once it is expressed—an elegant mechanism for rapid immune defense but also a double-edged sword.
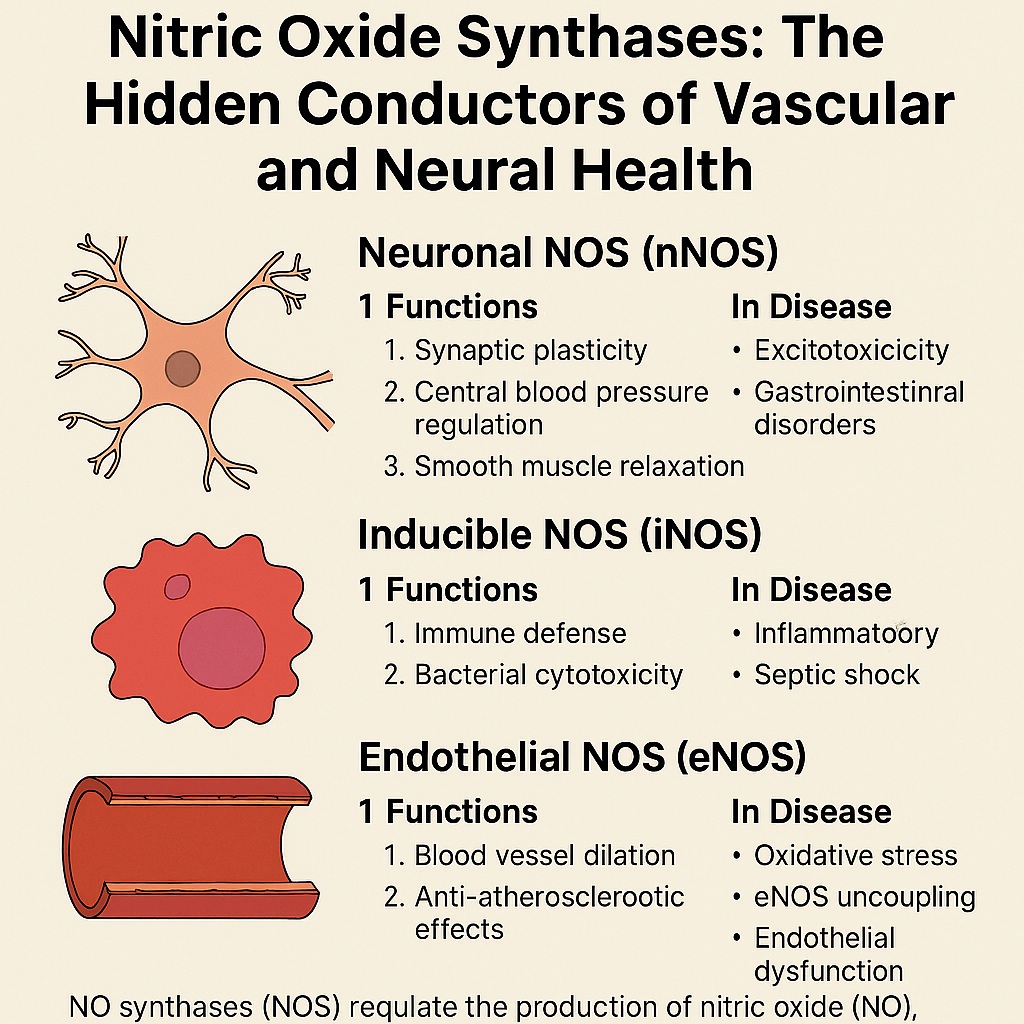
Neuronal Nitric Oxide Synthase (nNOS)
Neuronal NOS is widely distributed in the brain, spinal cord, adrenal glands, nitrergic nerves, and even skeletal muscle. It is constitutively expressed, and its activity depends on transient changes in calcium levels. In neurons, nNOS acts less as a switch for acute neurotransmission and more as a modulator of long-term plasticity. It influences learning, memory, and neurogenesis through retrograde signaling at synapses.
nNOS-derived NO also regulates central blood pressure. When inhibited in the brainstem, blood pressure rises; when hyperactivated, hypotension ensues. This delicate balance underscores how intimately the nervous system and vascular system are intertwined.
In the periphery, nitrergic nerves releasing NO from nNOS control smooth muscle relaxation. This is especially crucial in the corpus cavernosum, where NO triggers erection via cyclic GMP signaling. Here lies the rationale for phosphodiesterase-5 inhibitors—sildenafil, vardenafil, tadalafil—which rely on residual nNOS activity to sustain erections. Interestingly, the same mechanism underpins their use in pulmonary hypertension, since PDE5 is abundant in pulmonary arteries.
But nNOS is not always a hero. Hyperactivation contributes to excitotoxic neuronal death in stroke, where excessive calcium influx leads to overwhelming NO production, peroxynitrite formation, and mitochondrial injury. Likewise, inappropriate NO release in the gastrointestinal tract may underlie motility disorders. Thus, nNOS embodies the principle: in moderation, life; in excess, pathology.
Inducible Nitric Oxide Synthase (iNOS)
Unlike its neuronal and endothelial cousins, iNOS is not constitutively active. It is summoned in the heat of immune battle—induced by bacterial lipopolysaccharide, pro-inflammatory cytokines, or viral infection. Once expressed, iNOS churns out vast quantities of NO, independent of calcium.
This flood of NO confers cytotoxic power. By binding to iron centers of enzymes, NO disrupts energy metabolism, DNA synthesis, and mitochondrial respiration in pathogens. Macrophages use this weapon to kill Mycobacterium tuberculosis or Leishmania parasites. Hepatocytes can deploy iNOS to destroy malaria sporozoites. Even endothelial cells, under cytokine stimulation, can use NO to lyse tumor cells.
Yet, the destructive potential of iNOS is indiscriminate. Excess NO can spill into surrounding tissue, forming peroxynitrite and causing collateral damage. Autoimmune diseases, inflammatory lesions, and transplant rejection often show high iNOS activity. In the brain, activated microglia expressing iNOS contribute to neurodegeneration, combining NO with superoxide to induce neuronal apoptosis.
In septic shock, iNOS is the villain in chief. Massive systemic induction of iNOS leads to uncontrolled vasodilation, hypotension, and organ failure. Notably, mice lacking iNOS are resistant to septic shock, highlighting the enzyme’s central role. Therapeutically, targeting iNOS remains a challenge—suppressing it too much risks impairing host defense, while leaving it unchecked leads to fatal hypotension.
Endothelial Nitric Oxide Synthase (eNOS)
If iNOS is the immune system’s blunt weapon, eNOS is the cardiovascular system’s guardian angel. Localized primarily in endothelial cells, eNOS-derived NO maintains vascular homeostasis. It dilates blood vessels, inhibits platelet aggregation, prevents leukocyte adhesion, suppresses smooth muscle proliferation, and regulates vascular remodeling. Collectively, these actions make NO an anti-atherosclerotic principle.
Regulation of eNOS is nuanced. Beyond calcium-calmodulin activation, eNOS responds to mechanical forces like shear stress, which phosphorylate the enzyme and sustain NO release. Hormones such as estrogen and growth factors like VEGF also stimulate eNOS via kinase pathways. Conversely, caveolin-1 acts as a brake, inhibiting eNOS when bound. Heat shock protein 90, on the other hand, promotes its activation.
eNOS is also implicated in angiogenesis and mobilization of endothelial progenitor cells, critical for tissue repair and ischemic recovery. In experimental models, absence of eNOS impairs new vessel formation and fetal lung development. Enhancing eNOS activity pharmacologically or genetically improves neovascularization—a promising avenue in regenerative medicine.
Unfortunately, eNOS is vulnerable. Cardiovascular risk factors—hypertension, diabetes, smoking, hyperlipidemia—induce oxidative stress that uncouples eNOS, turning it into a superoxide generator. Loss of NO bioavailability contributes to endothelial dysfunction, the earliest step in atherosclerosis. Elevated levels of asymmetric dimethylarginine (ADMA), an endogenous eNOS inhibitor, exacerbate this problem.
NOS Uncoupling: When Protectors Become Villains
Uncoupling represents the dark side of NOS biology. Under oxidative stress, cofactors like BH4 are oxidized, and eNOS begins producing superoxide instead of NO. This not only eliminates the protective vasodilator but actively promotes vascular injury. Similar effects occur when L-arginine is depleted, when arginase competes excessively, or when ADMA accumulates.
Another emerging mechanism is S-glutathionylation of eNOS, a redox-sensitive modification that impairs NO synthesis and promotes superoxide release. In hypertensive models, reversing S-glutathionylation restores vasodilation, hinting at therapeutic opportunities.
The clinical consequences are vast: hypertension, atherosclerosis, ischemic heart disease, and diabetic vasculopathy are all tied to impaired eNOS coupling. Restoring balance is therefore a cornerstone of modern cardiovascular therapy.
Pharmacological Modulation of NOS
Interestingly, several conventional drugs improve endothelial function by influencing NOS.
- Renin–angiotensin system inhibitors (ACE inhibitors, angiotensin receptor blockers, renin inhibitors) reduce oxidative stress, increase BH4 availability, and restore eNOS activity. By curbing NADPH oxidase activity, they lower superoxide burden. Some, like losartan, directly enhance cofactor synthesis.
- Statins—famous for lowering cholesterol—also boost eNOS expression, reduce caveolin inhibition, and activate Akt pathways. They enhance BH4 levels and suppress NADPH oxidase, thereby preventing eNOS uncoupling. Their pleiotropic benefits—plaque stabilization, reduced thrombosis, and improved vascular tone—may owe as much to NO as to cholesterol reduction.
- Phosphodiesterase-5 inhibitors, though not directly acting on NOS, depend on intact nNOS and eNOS function for efficacy. By blocking cGMP degradation, they prolong NO signaling, whether in penile erection or pulmonary hypertension.
The therapeutic message is clear: supporting NOS function, especially eNOS, is central to cardiovascular protection.
Conclusions
Nitric oxide synthases exemplify biological duality. nNOS refines neural circuits and regulates blood pressure but may fuel excitotoxicity. iNOS defends against infection but can kill host tissue and trigger shock. eNOS safeguards vascular health but becomes destructive under oxidative stress.
Together, they illustrate how the same molecule—NO—can sustain life or drive disease depending on context. Understanding their regulation, vulnerabilities, and interactions with pharmacological agents is essential for modern medicine.
In the clinic, strategies that preserve eNOS coupling and limit iNOS overactivation hold particular promise. As our therapeutic arsenal expands, targeting the subtle regulatory levers of NOS may allow us to harness NO’s protective potential while taming its destructive power.
FAQ
1. Why is nitric oxide so important in cardiovascular health?
Nitric oxide maintains vascular tone, prevents clot formation, inhibits inflammation, and suppresses smooth muscle proliferation. Loss of NO, particularly from dysfunctional eNOS, contributes to endothelial dysfunction and atherosclerosis.
2. How do common drugs like statins or ACE inhibitors influence nitric oxide?
Beyond their primary targets, these drugs restore eNOS function, reduce oxidative stress, and increase NO bioavailability. This partly explains their cardiovascular benefits beyond cholesterol or blood pressure reduction.
3. Can boosting nitric oxide directly (e.g., supplements) improve health?
While L-arginine or BH4 supplementation can enhance NO production in some contexts, results are inconsistent. Targeted pharmacological approaches that restore NOS coupling and limit oxidative stress appear more effective than indiscriminate NO boosting.