
Heart failure rarely begins with drama. It begins quietly—an ejection fraction that slips below normal, a ventricle that remodels, a left atrium that enlarges—while the patient still insists they feel “fine.” This stage, commonly called preclinical systolic dysfunction (PSD) and aligned with ACC/AHA Stage B heart failure, is the calm before the storm: measurable left ventricular systolic dysfunction without the classic signs and symptoms of heart failure. The problem is not that nothing is happening. The problem is that enough is happening to set the stage for future decompensation.
One major driver of progression from PSD to symptomatic heart failure is impaired cardiorenal adaptation to volume load. In plain language: the heart and kidneys stop responding to extra salt and fluid the way they should. The kidneys fail to excrete sodium efficiently; the circulation holds on to volume; and the patient’s first “heart failure hospitalization” may be the first time anyone realizes the system has been drifting toward failure for years. The study you provided is built on this premise: if we can rescue the impaired cardiorenal response early, we may reduce the odds that PSD becomes overt heart failure.
To do that, the investigators focused on a pathway that connects cardiac function, vascular tone, and renal handling of sodium: the natriuretic peptide–cGMP system. B-type natriuretic peptide (BNP) stimulates cyclic GMP (cGMP), promoting vasodilation and natriuresis. But cGMP is degraded by phosphodiesterase type 5 (PDE5), and PDE5 upregulation has been implicated in impaired cGMP signaling in heart failure states. The therapeutic idea sounds elegant: combine PDE5 inhibition (to prevent cGMP breakdown) with exogenous BNP (to generate more cGMP), then challenge the system with volume expansion and see if the heart and kidneys behave more normally.
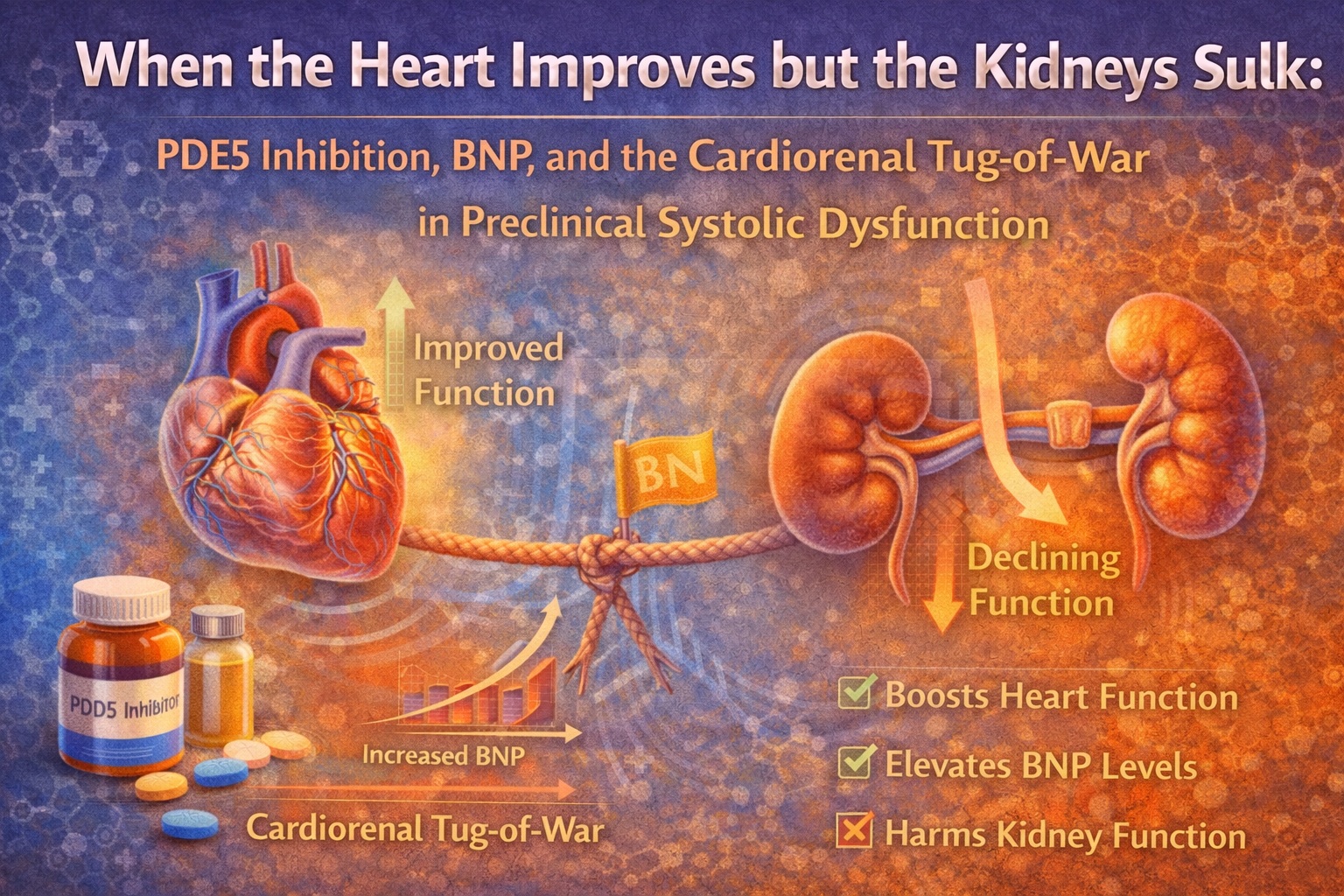
The twist—and the clinical lesson—is that physiology rarely rewards elegance without charging a fee. In this first-in-human trial, the combination improved cardiac function yet worsened renal function in response to acute saline loading. In other words: the heart said “thank you,” and the kidneys replied, “absolutely not.”
Why PSD Is the Perfect Place to Intervene—and the Worst Place to Be Overconfident
PSD is common in the community and is associated with higher rates of progression to symptomatic heart failure and increased mortality. The paper cites population data suggesting PSD affects a meaningful proportion of adults, and progression rates from PSD to overt heart failure are often quoted in the range of several percent per year—high enough to matter, low enough that many patients and clinicians underestimate it.
The danger in PSD is not only reduced contractility. It’s the loss of reserve. A person with PSD may appear stable until a stressor—salt load, infection, uncontrolled hypertension, atrial arrhythmia—tips them into congestion. That tipping point often involves the kidneys. When the kidneys cannot increase sodium excretion appropriately, fluid accumulates and symptoms follow. The authors note that impaired renal response to acute volume expansion has been documented in PSD, including paradoxically reduced renal cGMP activation and blunted natriuresis during saline loading compared with normal subjects.
This makes PSD an attractive therapeutic target: intervene early, before repeated congestion cycles and neurohormonal activation entrench disease. It also makes PSD a dangerous target for overinterpretation because improvements in a single marker (like ejection fraction) may not reflect a stable improvement in system-level behavior. The heart and kidneys operate as a coupled network. Helping one component at the expense of the other can be clinically counterproductive—especially if the long-term endpoint is “fewer decompensations,” not “nicer echocardiograms.”
This trial was designed around that systems concept. Instead of measuring long-term hospitalization outcomes (which would require large populations and long follow-up), it used a controlled physiologic stress test—acute saline volume expansion—to see whether combined therapy restores healthier cardiorenal adaptation in PSD with renal dysfunction.
The Core Hypothesis: “Boost cGMP and the System Will Behave Better”
BNP and related natriuretic peptides are not merely biomarkers. They are hormones with direct physiologic actions: vasodilation, natriuresis, and modulation of cardiac filling pressures. Their second messenger, cGMP, drives many of these effects. In heart failure and preclinical dysfunction, the natriuretic peptide system may be relatively insufficient for the degree of cardiovascular stress—sometimes described as a state where there is “too little of a good thing.”
PDE5 degrades cGMP. Experimental heart failure models have reported PDE5 upregulation, potentially contributing to impaired cGMP signaling and reduced renal responsiveness to natriuretic peptides. In animal studies, combining PDE5 inhibition with exogenous BNP enhanced cardiorenal responses more than either approach alone, suggesting synergy: BNP generates cGMP, and PDE5 inhibition prevents its breakdown. The authors highlight that while PDE5 inhibitors are clinically used (erectile dysfunction, pulmonary hypertension) and recombinant BNP (nesiritide) has been used in acute decompensated heart failure, the combination’s acute cardiorenal effects in humans had not been tested before this work.
The study’s objective was therefore crisp: in people with PSD and renal dysfunction, does tadalafil (PDE5 inhibitor) + subcutaneous BNP improve the cardiorenal response to acute volume expansion compared with tadalafil plus placebo?
If you like your science with a bit of drama, the answer is: yes for the heart, no for the kidneys, and the difference matters.
Study Design: Controlled Physiology, Not a “Real-World” Trial—and That’s the Point
This was a randomized, double-blinded, placebo-controlled crossover study conducted at Mayo Clinic. The crossover design matters: each participant served as their own control across two visits, reducing inter-individual variability in renal function, cardiac remodeling, and neurohormonal tone.
The population included adults with:
- Left ventricular ejection fraction (LVEF) <45%
- No current or previous diagnosis of heart failure
- Not on loop diuretics
- Renal dysfunction with estimated GFR between 30 and 90 ml/min
- Adequate functional capacity (6-minute walk >450 m without mechanical limitation)
A total of 25 were enrolled; 21 completed randomization and both crossover visits. Baseline characteristics show a cohort that looks like real-world Stage B disease: mean age ~67, BMI ~29, high prevalence of coronary artery disease (67%) and prior MI (33%), and widespread use of ACE inhibitor/ARB (86%) and beta-blocker (90%). Their baseline BNP and ANP were mildly elevated, while aldosterone and angiotensin II were within normal range—consistent with PSD rather than symptomatic heart failure with major RAAS activation.
The protocol was meticulous. Participants were stabilized on a low-salt diet (120 mEq/day), hydrated to ensure urine flow, and underwent renal clearance measurements using iothalamate and para-aminohippurate (classic techniques for GFR and renal plasma flow). They received either tadalafil 5 mg + subcutaneous placebo or tadalafil 5 mg + subcutaneous BNP 10 μg/kg, then underwent acute volume expansion with normal saline at 0.25 ml/kg/min for 1 hour. Echo and renal/humoral measures were obtained at baseline and after volume expansion.
This was not meant to simulate daily clinical life. It was meant to isolate physiology under controlled stress—exactly the right approach when you want to learn what a pathway intervention actually does to an integrated system.
What Happened With Tadalafil Alone: A Reasonable Renal Response, Minimal Cardiac Gain
When participants received tadalafil + placebo, acute volume expansion produced expected volume-related changes. LV end-diastolic volume (LVEDV) increased (a normal response to filling), and right ventricular systolic pressure increased modestly. LVEF did not change significantly. Neurohormonally, ANP and plasma cGMP increased, aldosterone decreased, while BNP and angiotensin II remained unchanged.
The renal response in this arm looked like what you would hope to see during a saline load: renal plasma flow increased, urine flow increased, and sodium excretion increased. GFR trended upward. In Table 2, renal plasma flow rose from ~303 to ~364 ml/min, urine flow from ~4.4 to ~7.1 ml/min, and sodium excretion from ~159 to ~245 mEq/min (all statistically significant except the GFR trend).
In other words, tadalafil alone did not create a dramatic improvement in cardiac function in the face of a volume challenge—but the kidneys still did their job. This is a crucial baseline observation because it frames the combination results: the kidney deterioration seen with BNP addition was not simply “PSD physiology under volume load.” It was a differential response linked to the combination intervention.
If you were hoping for a story where one elegant combination fixes everything, this is where you should stop hoping. But if you prefer stories that sound like real medicine, the plot is just starting.
What Happened With Tadalafil + BNP: Better Cardiac Performance, Worse Renal Handling
With tadalafil + subcutaneous BNP, the systemic and organ-level responses shifted noticeably.
Hemodynamically, systolic blood pressure fell significantly during the intervention and volume expansion (Table 2 shows systolic BP dropping from ~125 to ~112 mmHg, diastolic from ~69 to ~60 mmHg). Heart rate rose modestly.
Cardiac function improved in ways that are hard to ignore. With volume expansion, the combination arm showed:
- Increased LVEF (from ~38.8% to ~43.1%, p<0.001)
- Reduced LVEDV (from ~199 to ~177 ml, p=0.005)
- Reduced LVESV (from ~128 to ~101 ml, p<0.001)
- Reduced left atrial volume index (LAVI) (from ~86 to ~76 ml/m², p=0.017)
This pattern—higher EF with smaller volumes and reduced LA size index—suggests improved systolic performance and lower filling pressures in response to volume challenge, rather than simple volume accumulation. The authors reinforce this interpretation by noting that ANP decreased in the BNP arm relative to placebo, suggesting reduced cardiac filling pressure response to volume expansion.
Now the renal side: it went the other way. In the tadalafil+BNP arm, during volume expansion there was:
- Decreased renal plasma flow (RPF) (from ~334 to ~259 ml/min, p=0.022)
- Decreased GFR (from ~79 to ~68 ml/min, p=0.015)
- No increase in urine flow
- No significant increase in sodium excretion
This is the key clinical paradox: the kidneys failed to respond appropriately to a saline load, despite the cGMP pathway being strongly activated. Urinary cGMP excretion increased dramatically (from ~853 to ~3,420 pmol/min, p<0.001), and plasma cGMP rose sharply as well.
If you only looked at cGMP, you would predict better natriuresis. If you only looked at EF, you might congratulate yourself. But if you look at renal plasma flow and GFR, you start worrying about precisely the thing that drives decompensation: impaired volume handling.
The visual summary in Table 2 (page 6) captures the split: improved cardiac metrics versus worsened renal metrics under the same volume expansion stress.
Why Would BNP + PDE5 Inhibition Hurt the Kidney When cGMP Is High?
The authors argue the likely culprit is blood pressure and renal perfusion pressure. The combination produced a greater drop in systolic BP than tadalafil alone. Lower systemic pressure can translate into reduced renal perfusion pressure. When renal perfusion falls, autoregulatory mechanisms try to maintain GFR, but they can fail if vasodilatory/vasoconstrictive balance is overwhelmed—particularly in patients with pre-existing renal dysfunction.
From classic renal physiology: reduced perfusion pressure lowers sodium delivery to the nephron (especially the loop of Henle), increasing sodium reabsorption and reducing natriuresis. This can blunt the diuretic response to volume loading and contribute to volume retention—the very state BNP was supposed to help prevent.
The paper also explored whether the BP drop directly correlated with renal deterioration and did not find a significant association—but the authors note the sample size was small, so absence of correlation is not strong evidence of absence of effect. They also performed subgroup analyses by estimated GFR (<60 vs ≥60 ml/min). Interestingly, those with higher baseline eGFR appeared to have statistically worsened renal outcomes in the BNP arm versus placebo, though small numbers prevented firm conclusions, and the authors explicitly call for larger studies.
Clinically, the most important message is this: you can enhance a signaling pathway and still lose organ function if perfusion pressure drops too far. Biochemistry does not override hemodynamics. The kidney is not impressed by beautiful second messengers when it is under-perfused.
Safety Signals: Not Catastrophic, but Not “Nothing Happened” Either
Adverse events were minimal with tadalafil + placebo. In the tadalafil + BNP arm, a small number of participants experienced symptoms consistent with the intervention’s hemodynamic effect: nausea/vomiting (10%), transient chest discomfort (5%), and hypotension (5%) that resolved with saline infusion.
These are not shocking events in a controlled research unit setting, but they matter for translation. They signal that adding exogenous BNP in this population can produce meaningful systemic effects, including hypotension—exactly the mechanism implicated in renal function worsening. In real-world outpatient practice, hypotension episodes can be missed, underreported, or interpreted as “just dehydration,” while the kidney pays the price.
This is also why the authors emphasize the need to explore different dosing strategies. The tested doses were single-dose choices: tadalafil 5 mg and subcutaneous BNP 10 μg/kg. They explicitly suggest that lower, non-hypotensive doses might preserve cardiac benefit without harming renal function, but that hypothesis remains to be tested.
Clinical Meaning: What This Teaches About Stage B Disease and “Fixing” Cardiorenal Dysfunction
The study’s greatest contribution is not a ready-to-prescribe regimen. It is a demonstration of differential organ response in an early heart failure state: cardiac function can improve acutely while renal function worsens under stress.
That matters because PSD management is often framed as preventing progression through standard therapies—blood pressure control, ACEi/ARB, beta-blockers, lifestyle, ischemia management. This work suggests a complementary dimension: the capacity to handle volume challenges may be impaired before symptoms appear, and interventions targeting natriuretic peptide signaling can shift cardiac and renal performance in opposite directions.
It also has implications for therapies that modulate the natriuretic peptide system more broadly. The discussion mentions neprilysin inhibition (e.g., sacubitril/valsartan), which enhances natriuretic peptide signaling by reducing degradation and improves outcomes in symptomatic HFrEF. In PSD, where natriuretic peptide levels may be only mildly elevated and RAAS may not be strongly activated, the balance of benefit and harm might differ—especially in patients with renal dysfunction.
Finally, it offers a clinical caution: if you attempt to “push” vasodilation and natriuretic peptide biology in a patient with borderline kidney perfusion, you may win the echo and lose the kidney. In medicine, that is rarely a good trade.
What to Do Next: Practical Research Directions Without Pretending We’re Already Done
The authors themselves are disciplined: they emphasize that this was an acute physiologic study and may not reflect long-term effects. They also highlight that only one dosing strategy was tested and that different populations (e.g., hypertensive patients) might respond differently.
From a translational viewpoint, the next steps are obvious:
- Explore dose-finding to identify non-hypotensive combinations that preserve renal perfusion
- Test longer-term regimens to see whether the acute renal signal translates into chronic harm or adaptation
- Identify phenotypes within PSD (renal function strata, BP profiles, NP levels) that predict benefit versus risk
If the goal is prevention of symptomatic heart failure, the endpoint must ultimately be clinical outcomes, not only short-term changes in cGMP, EF, or urine flow. But physiology studies like this one are essential to avoid large, expensive outcome trials built on assumptions that are physiologically incorrect.
Conclusion: The “Same Pathway” Does Not Mean the Same Outcome in Different Organs
In adults with preclinical systolic dysfunction and renal dysfunction, pretreatment with tadalafil plus subcutaneous BNP before acute saline volume expansion improved cardiac function—higher LVEF and reduced cardiac volumes and left atrial volume index—while simultaneously worsening renal response, with reductions in renal plasma flow, GFR, and urine output response compared with tadalafil alone. The combination strongly activated cGMP in plasma and urine, yet the renal physiology still deteriorated, likely influenced by hypotension and reduced renal perfusion.
The clinical lesson is simple and slightly humbling: improving heart performance is not the same as restoring cardiorenal resilience. In PSD, the kidneys are not passive observers. They are co-authors of the patient’s future heart failure story—and they may refuse to cooperate with a therapy that looks excellent on paper and slightly less excellent on perfusion pressure.
FAQ
1) What is preclinical systolic dysfunction (PSD), and why should we treat it if patients feel fine?
PSD is systolic dysfunction (reduced LVEF) without symptoms of heart failure, corresponding to ACC/AHA Stage B. It carries a meaningful risk of progression to symptomatic heart failure and adverse outcomes, so early identification and targeted prevention strategies are clinically important.
2) Why did tadalafil + BNP improve cardiac function but worsen renal function during volume expansion?
The combination increased cGMP signaling and improved echocardiographic measures, but it also produced a greater drop in blood pressure, likely reducing renal perfusion pressure. In patients with renal dysfunction, reduced perfusion can lower RPF and GFR and blunt urine/sodium excretion responses even when cGMP levels are high.
3) Does this mean we should use tadalafil plus BNP in Stage B heart failure?
Not yet. This was an acute physiologic study testing a single dosing strategy. It suggests potential cardiac benefit but raises concern for renal harm, and the authors call for additional studies—especially dose optimization and longer-term outcome evaluation—before clinical adoption.
