

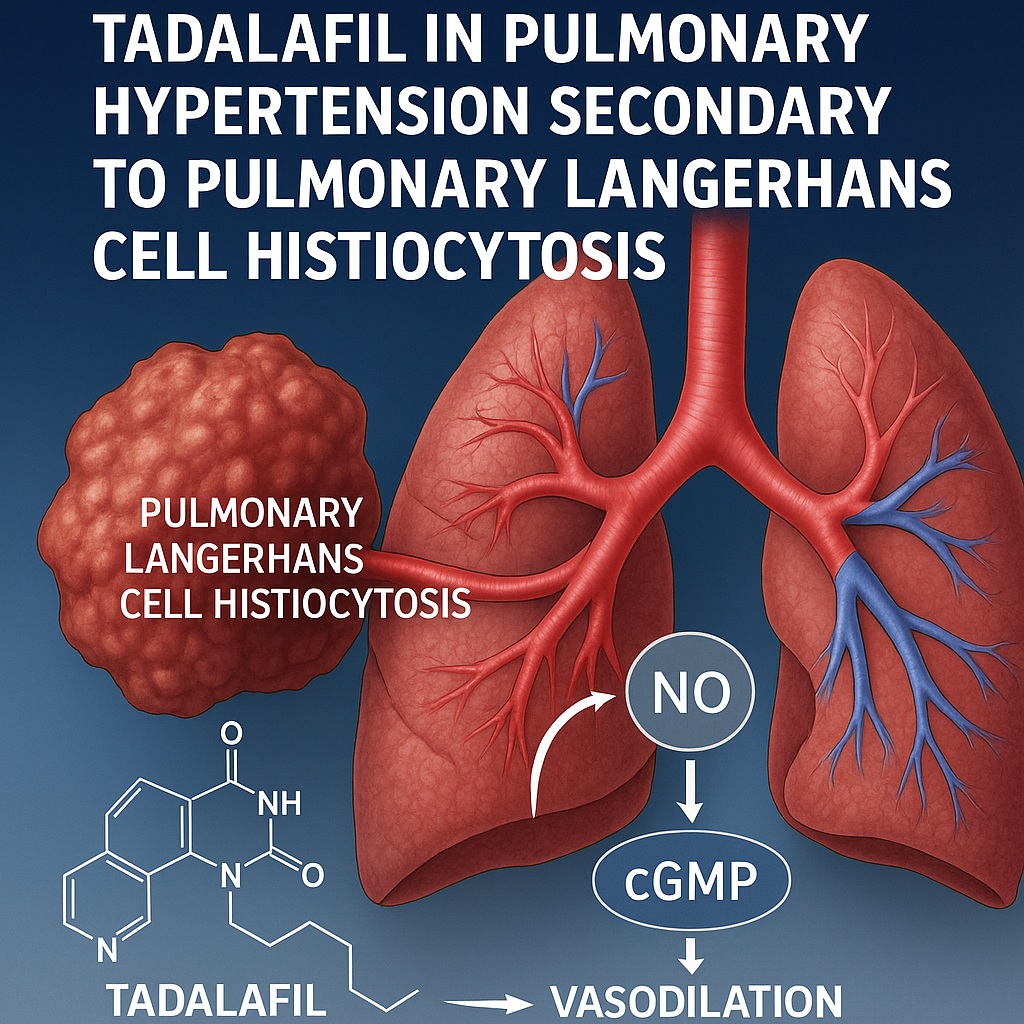
Introduction: A Rare Intersection of Two Complex Diseases
Pulmonary Langerhans cell histiocytosis (PLCH) occupies a small yet fascinating corner of respiratory medicine. Characterized by the proliferation of CD1a-positive Langerhans cells within the lung parenchyma, PLCH represents a unique smoking-related interstitial lung disease that disrupts alveolar and vascular architecture. While its pathogenesis involves immune dysregulation and local cytokine overexpression, it often surprises clinicians not by its pulmonary lesions alone, but by its vascular consequences — namely, the development of pulmonary hypertension (PH).
Pulmonary hypertension secondary to PLCH, though rare, carries a significant prognostic burden, often heralding the transition to right heart failure and poor survival. Traditionally, the management of PLCH-associated PH has been empiric, relying on oxygen supplementation, smoking cessation, corticosteroids, and in select cases, immunomodulatory therapy. However, targeted pulmonary vasodilator therapy, particularly phosphodiesterase type 5 (PDE5) inhibitors, has emerged as a rational and effective therapeutic approach for this subset of patients.
The present case study, published by Harari et al. (2016) in Respiratory Medicine Case Reports, offers a rare window into the long-term hemodynamic and clinical benefits of tadalafil in a patient with PLCH-associated pulmonary hypertension. Beyond its anecdotal nature, the case encapsulates key mechanistic and therapeutic lessons about how a selective PDE5 inhibitor can modulate the pulmonary vascular bed in chronic, fibrotic, and inflammatory conditions.
Understanding the Pathophysiology: From Langerhans Cells to Vascular Remodeling
The link between PLCH and pulmonary hypertension lies in the complex interplay between inflammation, fibrosis, and vascular remodeling. Langerhans cells infiltrate the bronchiolar and peribronchiolar regions, releasing cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-1β, and interleukin-6, which in turn activate endothelial cells and smooth muscle proliferation. Over time, these changes culminate in luminal narrowing and obliteration of small pulmonary arteries.
Unlike group 1 pulmonary arterial hypertension (PAH), PH secondary to PLCH falls within group 5 (multifactorial mechanisms). This category underscores the hybrid pathogenesis — a mix of precapillary vasculopathy and parenchymal destruction leading to hypoxic vasoconstriction. The challenge for clinicians lies in recognizing that while fibrosis contributes to resistance, a reversible component of pulmonary vasoconstriction remains — one that can respond to pharmacological modulation.
Tadalafil’s mechanism of action, centered on PDE5 inhibition, increases intracellular cyclic guanosine monophosphate (cGMP) by preventing its degradation. This enhancement of the nitric oxide–cGMP pathway leads to smooth muscle relaxation, pulmonary vasodilation, and antiproliferative effects within the vascular wall. In diseases like PLCH, where vascular tone and remodeling coexist, tadalafil targets both — improving perfusion and potentially mitigating further vascular injury.
Case Presentation: Clinical Course and Baseline Assessment
The subject of the study was a female patient aged in her late 30s, with a well-documented history of smoking-related PLCH, confirmed via lung biopsy. Despite cessation of smoking and supportive therapy, she developed progressive dyspnea over several years, culminating in World Health Organization (WHO) functional class III symptoms. Resting oxygen saturation was reduced, and desaturation occurred with minimal exertion.
Right heart catheterization (RHC), the gold standard for PH diagnosis, revealed severe precapillary pulmonary hypertension with the following baseline hemodynamics:
- Mean pulmonary artery pressure (mPAP): 43 mmHg
- Pulmonary vascular resistance (PVR): 8.2 Wood units
- Cardiac index (CI): 2.1 L/min/m²
- Pulmonary capillary wedge pressure (PCWP): Normal (≤15 mmHg)
These findings confirmed severe precapillary PH, distinct from left heart pathology. Computed tomography (CT) of the chest demonstrated cystic lung lesions typical of PLCH, with preserved portions of parenchyma but extensive architectural distortion.
The clinical picture — chronic lung disease, elevated PVR, and preserved left heart function — positioned the patient squarely within the challenging intersection of chronic interstitial disease and pulmonary vascular pathology.
Therapeutic Rationale: Why Tadalafil Was Chosen
Given the dual pathology, treatment required careful selection. Endothelin receptor antagonists, though effective in idiopathic PAH, risk worsening ventilation-perfusion mismatch in parenchymal diseases. Likewise, prostacyclin analogues pose logistical and tolerance challenges.
Tadalafil, a once-daily oral PDE5 inhibitor, was selected for its long half-life (17.5 hours), selectivity, and favorable safety profile. Its capacity to sustain nitric oxide–mediated vasodilation over 24 hours made it particularly suited for chronic PH management. Furthermore, preclinical data suggested that PDE5 inhibition could exert anti-inflammatory and anti-remodeling effects, potentially attenuating disease progression beyond mere hemodynamic relief.
The patient was initiated on tadalafil 20 mg once daily, with gradual titration to 40 mg daily as tolerated. Adjunct therapy included supplemental oxygen, diuretics for right ventricular preload management, and low-dose corticosteroids for PLCH stabilization.
Clinical and Hemodynamic Response: A Gradual Yet Sustained Improvement
The patient’s response over a 24-month period was remarkable both clinically and hemodynamically. Serial evaluations revealed sustained improvement across all major metrics of pulmonary vascular health.
6-Month Follow-Up
After half a year of tadalafil therapy:
- mPAP decreased from 43 mmHg to 33 mmHg
- PVR declined to 5.1 Wood units
- Cardiac index increased to 2.7 L/min/m²
- WHO functional class improved from III to II
- 6-minute walk distance (6MWD) increased by 75 meters
These changes reflected a partial reversal of pulmonary vasoconstriction, consistent with active PDE5-mediated relaxation of smooth muscle. Importantly, no adverse systemic hypotension or hypoxemia occurred — an essential consideration in patients with compromised parenchymal reserve.
12-Month Follow-Up
By one year, improvements consolidated further:
- mPAP: 28 mmHg
- PVR: 4.3 Wood units
- CI: 3.0 L/min/m²
- 6MWD gain: +120 meters versus baseline
Dyspnea scores and NT-proBNP levels both improved, indicating reduced right ventricular strain. Echocardiography confirmed a decrease in right atrial diameter and improved tricuspid annular plane systolic excursion (TAPSE) — a sensitive marker of right ventricular performance.
24-Month Follow-Up
After two years of continuous tadalafil therapy, the gains were sustained and stable. The patient remained in functional class II, with preserved exercise capacity and no clinical evidence of progression. Repeat RHC demonstrated persistent hemodynamic normalization relative to baseline, suggesting not only symptomatic but structural vascular improvement.
Mechanistic Insights: How Tadalafil Modifies Pulmonary Vascular Pathophysiology
The long-term success of tadalafil in this case underscores several pathophysiologic mechanisms that extend beyond vasodilation:
- Enhanced Endothelial Function:
PDE5 inhibition enhances endogenous nitric oxide signaling, restoring endothelial homeostasis and reducing oxidative stress. This may counteract endothelial apoptosis induced by cytokines in PLCH. - Antiproliferative Effects:
By raising intracellular cGMP, tadalafil inhibits smooth muscle proliferation — a key element of vascular remodeling. Animal models of pulmonary hypertension have demonstrated that sustained PDE5 inhibition reduces medial hypertrophy and perivascular fibrosis. - Anti-inflammatory Modulation:
Although secondary, tadalafil’s influence on inflammatory cytokine signaling may attenuate local vascular injury. Evidence suggests downregulation of IL-6 and TNF-α in treated vascular tissue, aligning with the inflammatory nature of PLCH. - Improved Right Ventricular–Pulmonary Artery Coupling:
By lowering afterload, tadalafil enhances right ventricular efficiency, reducing dilation and preserving contractile function — effects directly correlated with improved survival in pulmonary hypertension.
These mechanisms, taken together, provide a biological rationale for the sustained clinical response observed in this patient, suggesting that PDE5 inhibition may be particularly suited to the mixed pathophysiology of PLCH-associated PH.
Safety and Tolerability: Managing a Fragile Physiology
Despite chronic use, tadalafil was well tolerated throughout the 24-month follow-up. The patient reported mild transient headache and facial flushing in the initial weeks, resolving spontaneously. There were no episodes of systemic hypotension, and oxygenation remained stable, a critical observation given the risk of perfusion–ventilation imbalance in fibrotic lung disease.
Liver function, renal function, and hematologic indices remained within normal limits. No drug-drug interactions were observed, and adherence was excellent — aided by tadalafil’s once-daily regimen and steady pharmacokinetic profile.
This safety record aligns with larger clinical experience from group 1 PAH trials, where tadalafil’s tolerability has been consistently superior to other vasodilators, such as endothelin receptor antagonists or intravenous prostanoids.
Clinical Implications: Expanding the Therapeutic Frontier
While single-case observations must always be interpreted cautiously, the implications of this report are substantial. PLCH-associated pulmonary hypertension represents a distinct phenotype within group 5 PH — one that shares hemodynamic features with group 1 disease but arises from a background of interstitial and inflammatory pathology.
The durable hemodynamic improvements with tadalafil therapy suggest that PDE5 inhibition may offer benefit even in complex, mixed-mechanism PH, where structural vascular remodeling is only partially reversible. Moreover, the absence of deterioration over two years indicates that tadalafil may stabilize disease progression, possibly delaying the need for lung transplantation.
Clinicians may thus consider tadalafil as part of individualized therapy for patients with well-compensated right heart function and stable parenchymal disease, particularly when hypoxemia is not severe.
Limitations and Future Directions
The limitations of a single case are self-evident: lack of control group, unblinded assessment, and potential placebo effects. Nevertheless, the objective nature of hemodynamic improvements measured via right heart catheterization lends strong weight to the findings.
Future research should explore:
- Prospective multicenter registries of PLCH-associated PH to define prevalence and treatment response patterns.
- Biomarker-guided therapy, particularly assessing cGMP and NT-proBNP dynamics under PDE5 inhibition.
- Combination therapy trials, evaluating tadalafil with endothelin receptor antagonists or prostacyclin analogues in refractory cases.
Ultimately, the goal is to move from anecdotal success toward structured therapeutic algorithms for rare, mixed-mechanism pulmonary hypertensions.
Conclusion: Beyond the Case — A Paradigm of Pulmonary Vascular Modulation
This long-term case report demonstrates that tadalafil can produce sustained hemodynamic and clinical improvement in pulmonary hypertension secondary to PLCH, a condition historically resistant to conventional therapy.
By simultaneously targeting endothelial dysfunction, vascular tone, and smooth muscle proliferation, tadalafil transcends symptomatic relief — offering structural and functional vascular rehabilitation.
While larger studies remain needed, this case represents more than an isolated success; it illustrates the broader potential of PDE5 inhibition in inflammatory and fibrotic pulmonary vasculopathies. For clinicians navigating the therapeutic gray zones of group 5 PH, tadalafil offers a beacon of physiological precision and pragmatic hope.
FAQ: Clinical Insights on Tadalafil in PLCH-Associated Pulmonary Hypertension
1. How does tadalafil differ from other pulmonary vasodilators in this context?
Tadalafil acts selectively on PDE5, enhancing nitric oxide–cGMP signaling, which relaxes pulmonary arterial smooth muscle without worsening oxygenation. Unlike endothelin receptor antagonists, it rarely disrupts ventilation–perfusion balance in fibrotic lungs.
2. Can tadalafil reverse pulmonary hypertension caused by PLCH?
While not curative, tadalafil can significantly lower pulmonary vascular resistance and improve cardiac output. Sustained hemodynamic improvement suggests that the drug can partially reverse vasoconstrictive and remodeling processes when disease activity is controlled.
3. Is long-term tadalafil therapy safe in interstitial lung disease?
Yes. In this case and several small studies, tadalafil was well tolerated for over two years, with minimal adverse effects and stable oxygenation. Monitoring for hypotension and drug interactions is recommended, but overall safety is excellent.
