
Understanding why some breast tumors succumb to chemotherapy while others remain stubbornly unresponsive is one of the most compelling puzzles in modern oncology. Doxorubicin—a cornerstone anthracycline used worldwide—exerts its antitumor effects primarily by inflicting double-strand DNA breaks. Yet breast cancer cells have evolved a complex interplay of epigenetic and post-transcriptional defense systems to neutralize this assault. Among these is RNA N⁶-methyladenosine (m⁶A), the most abundant internal modification on eukaryotic mRNA, which dynamically modulates RNA splicing, stability, and translation.
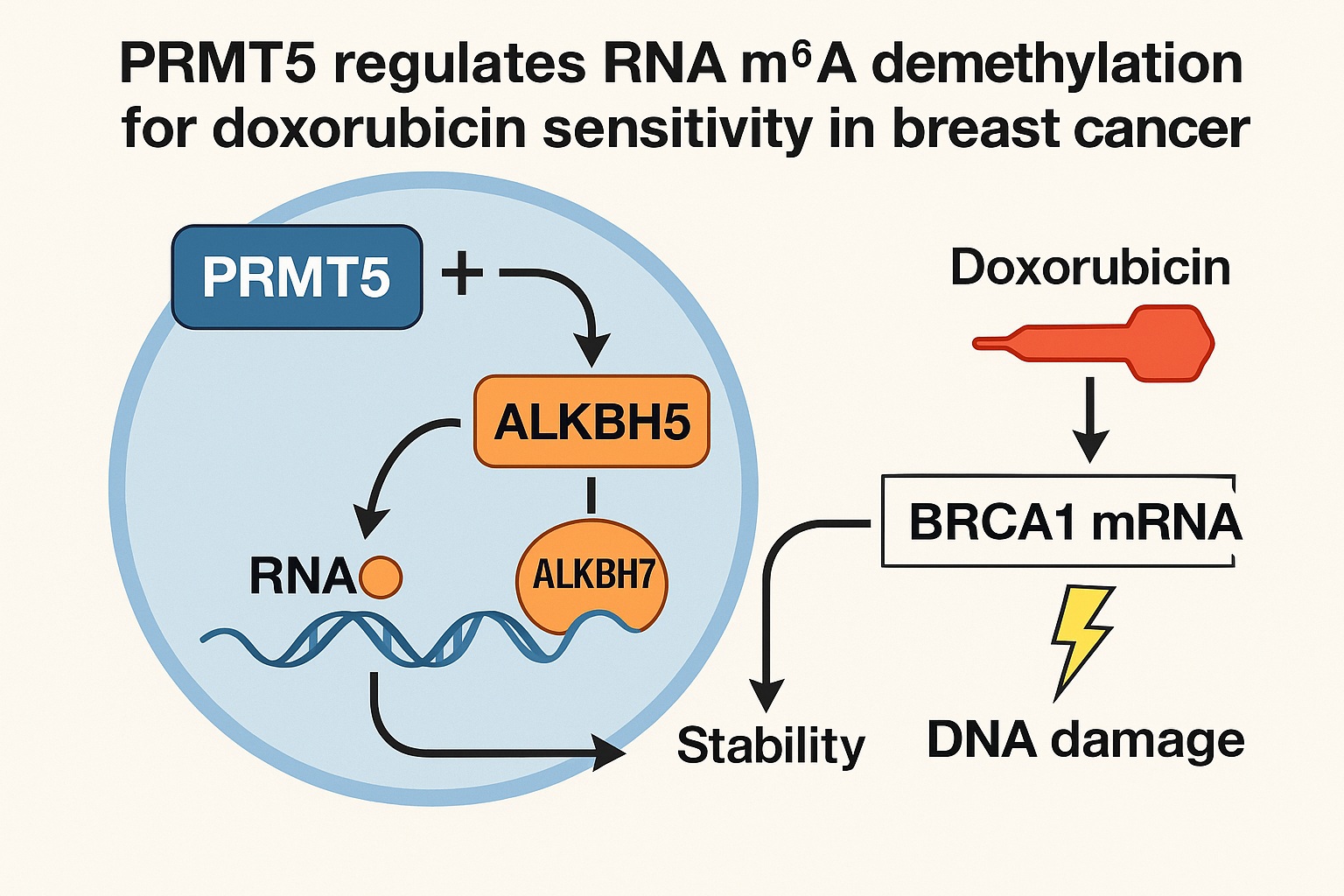
A recent investigation reveals that PRMT5, a protein arginine methyltransferase with well-established oncogenic activity, orchestrates a finely tuned resistance mechanism by regulating m⁶A demethylation of BRCA1 mRNA. This regulation occurs through an unexpected partnership between ALKBH5, a canonical nuclear m⁶A demethylase, and ALKBH7, a protein previously pigeonholed as a mitochondrial metabolic regulator. Even more strikingly, the study identifies tadalafil, a widely used PDE5 inhibitor, as a functional PRMT5 inhibitor capable of restoring doxorubicin sensitivity.
This article provides a comprehensive and highly readable explanation of this mechanism, grounded in rigorous molecular detail yet delivered with clarity and clinical relevance.
Doxorubicin, m⁶A Methylation, and the Initial Puzzle
Doxorubicin’s cytotoxicity is rooted in its ability to intercalate into DNA, inhibit topoisomerase II, and generate reactive oxygen species—all culminating in DNA double-strand breaks. Traditionally, the canonical response network to this damage has been framed in terms of DNA repair proteins, checkpoint kinases, and apoptotic pathways. But accumulating evidence suggests that epitranscriptomic responses—particularly m⁶A modifications—play a decisive role in modulating how cells cope with DNA injury.
In breast cancer samples exposed to neoadjuvant doxorubicin, global m⁶A levels are significantly elevated (see dot-blot assays on page 1). This increase is reproduced in breast cancer cell lines and patient-derived xenograft (PDX) models, where rising m⁶A levels closely parallel the accumulation of γ-H2AX, a hallmark of DNA damage. The response is both dose-dependent and time-dependent, emphasizing that m⁶A methylation is not a passive feature but an acute epitranscriptomic reaction to genotoxic stress.
However, this response is not uniform across all tumors. Breast cancer cells with high expression of PRMT5 display a blunted increase in m⁶A methylation following doxorubicin exposure. The implication is clear: PRMT5 interferes with the normal m⁶A-mediated DNA damage response, reducing doxorubicin efficacy and enabling tumor survival.
From a biological standpoint, this raises a critical question: How does a protein traditionally associated with arginine methylation of histones and spliceosomal proteins regulate an mRNA modification such as m⁶A?
PRMT5 as a Gatekeeper of RNA m⁶A and BRCA1 Stability
The search for downstream effects of PRMT5 overexpression reveals that it dampens doxorubicin-induced m⁶A methylation on numerous transcripts, but most prominently on BRCA1 mRNA. BRCA1 serves as the central architect of homologous recombination (HR), the most precise pathway for repairing DNA double-strand breaks.
A reduction in m⁶A on BRCA1 transcripts may initially appear counterintuitive, but the logic becomes clear when considering the relationship between m⁶A marks and mRNA turnover. m⁶A often signals transcripts for accelerated degradation. Reducing m⁶A on BRCA1 stabilizes the transcript, prolongs its availability, and ultimately bolsters DNA repair capability.
This mechanism is supported by multiple experimental observations:
- MeRIP-seq (page 2–3) demonstrates increased m⁶A peaks on BRCA1 after doxorubicin exposure, which are substantially diminished when PRMT5 is overexpressed.
- Luciferase assays with wild-type and m⁶A-site-mutated BRCA1 3’-UTR regions confirm that PRMT5-mediated effects depend on two specific m⁶A sites (at nucleotides 149 and 1268).
- RNA stability analysis using flavopiridol shows that PRMT5 preserves BRCA1 transcript stability even under doxorubicin stress.
- Functional assays—such as comet assays and γ-H2AX staining—demonstrate that reducing BRCA1 levels reverses PRMT5-induced chemoresistance.
Thus, PRMT5 acts as a molecular insurance policy for the cancer cell, ensuring that BRCA1-mediated repair continues even under genotoxic duress.
But the molecular question deepens: PRMT5 methylates arginine, not adenosine. So how does it regulate an RNA demethylation process? The answer lies in its unexpected influence on the cellular movement and stability of key m⁶A demethylases.
ALKBH7: A Surprising Link Between Arginine Methylation and RNA Demethylation
When searching for proteins physically interacting with PRMT5, ALKBH7 emerges as a leading candidate in mass spectrometry analyses (page 4–5). ALKBH7 has been historically categorized as a mitochondria-localized protein involved in metabolic stress and necrosis—a far cry from epitranscriptomic regulation.
Yet PRMT5 tightly binds ALKBH7 and catalyzes symmetric dimethylation at three arginine residues. Mutating these residues abrogates ALKBH7 methylation, reduces its protein stability, and enhances its degradation via the ubiquitin–proteasome system. This stability is essential, because ALKBH7 serves as a physical partner and chaperone for ALKBH5, the primary nuclear m⁶A demethylase.
Without PRMT5-mediated methylation, ALKBH7 is rapidly degraded, ALKBH5 fails to accumulate in the nucleus, and m⁶A demethylation becomes impaired.
The following experimental findings cement this relationship:
- Overexpressing ALKBH7 reduces global m⁶A levels, mimicking PRMT5 activity.
- Knocking down ALKBH7 reverses PRMT5-mediated demethylation and restores BRCA1 m⁶A accumulation.
- Nuclear–cytoplasmic fractionation (page 7) shows ALKBH5 translocates to the nucleus only when functional ALKBH7 is present.
- ALKBH7–ALKBH5 interactions are confirmed via co-immunoprecipitation.
This partnership indicates that PRMT5 does not demethylate RNA directly but modulates the machinery that performs the demethylation.
From a conceptual standpoint, it is a masterful strategy: reinforce DNA repair by stabilizing the very proteins that suppress m⁶A marks on BRCA1. The cancer cell becomes more adept at withstanding doxorubicin, enabling tumor survival and evolution.
Nuclear Trafficking of ALKBH5: The Determining Step in RNA m⁶A Dynamics
ALKBH5 has long been discussed as a predominantly nuclear enzyme, but this study reveals a far more dynamic reality. In untreated breast cancer cells, ALKBH5 resides both in the nucleus and cytoplasm. Under doxorubicin stress, its nuclear accumulation increases—but only in the presence of functional PRMT5–ALKBH7 signaling.
When ALKBH7 is silenced or its methylation sites mutated, ALKBH5 fails to translocate properly. This results in:
- Elevated global m⁶A levels
- Higher m⁶A enrichment at BRCA1 3’-UTR
- Reduced BRCA1 mRNA stability
- Increased doxorubicin-induced DNA damage
Thus, ALKBH5 nuclear localization becomes the fulcrum upon which doxorubicin sensitivity pivots. PRMT5, through ALKBH7, effectively decides whether ALKBH5 will rescue BRCA1 from m⁶A-mediated decay.
From an oncologic perspective, this mechanism explains clinical correlations observed in IHC analyses (page 8–9), where tumors with high PRMT5 expression show elevated nuclear ALKBH5, high ALKBH7 levels, and high BRCA1 expression. Not surprisingly, these tumors are also associated with poorer survival outcomes.
Repurposing Tadalafil as a PRMT5 Inhibitor: A Clinically Practical Strategy
One of the most compelling outcomes of this research is the identification of tadalafil, an FDA-approved PDE5 inhibitor, as a potent PRMT5 inhibitor. Virtual screening of 1813 approved drugs followed by molecular docking singled out tadalafil as a strong binder to the PRMT5 active site (page 10). This interaction was validated using surface plasmon resonance, in vitro methylation assays, and functional cellular assays.
Tadalafil effectively:
- Inhibits PRMT5-dependent arginine methylation
- Reduces ALKBH7 stability
- Blocks ALKBH5 nuclear translocation
- Restores m⁶A accumulation on BRCA1
- Reduces BRCA1 expression
- Sensitizes breast cancer cells to doxorubicin
In vitro, tadalafil synergizes with doxorubicin to suppress cell growth, particularly in BRCA1-wild-type breast cancer cells. In vivo, the combination dramatically reduces tumor burden in xenograft and PDX models (page 11–12).
These effects are abolished when PRMT5—not PDE5—is silenced, confirming that tadalafil’s anticancer function is PRMT5-dependent rather than related to its classical vasodilatory mechanism.
The practical implications are profound: because tadalafil is already clinically approved with a well-characterized safety profile, its repositioning as a chemosensitizer could proceed significantly faster than the development of novel PRMT5 inhibitors.
Implications for Clinical Practice and Future Oncology Therapeutics
This study reshapes our understanding of chemoresistance by illustrating how post-transcriptional RNA modifications influence DNA repair capacity. It also provides a translationally realistic therapeutic strategy that leverages drug repurposing to overcome a significant clinical dilemma.
Several aspects merit particular attention:
- Precision oncology potential: PRMT5 and ALKBH5 nuclear localization patterns could serve as biomarkers to predict doxorubicin response.
- Combination therapy strategies: Tadalafil may complement existing PARP inhibitors, as PRMT5 suppression sensitizes BRCA1-proficient tumors to DNA-damaging agents.
- Therapeutic window: Because PRMT5 plays roles in normal tissues, careful profiling will be needed to determine which patients are best suited for this approach.
- Epitranscriptomic therapeutics: This study underscores that targeting RNA modification machinery is becoming as important as targeting DNA repair pathways.
In the context of aggressive or treatment-refractory breast cancer, such mechanistic clarity is invaluable. Understanding how cancer cells manipulate m⁶A marks to survive chemotherapy allows researchers and clinicians to intervene with precision and creativity.
FAQ
1. Why does reducing m⁶A on BRCA1 make breast cancer cells more resistant to doxorubicin?
m⁶A marks often signal mRNA transcripts for degradation. Lower m⁶A levels on BRCA1 stabilize its mRNA, enabling higher BRCA1 protein production. Because BRCA1 is central to repairing DNA double-strand breaks, increased BRCA1 activity helps cancer cells rapidly repair doxorubicin-induced damage—thereby reducing chemotherapy efficacy.
2. How exactly does tadalafil enhance doxorubicin sensitivity?
Tadalafil inhibits PRMT5, preventing methylation and stabilization of ALKBH7. Without stable ALKBH7, ALKBH5 cannot translocate efficiently into the nucleus to demethylate m⁶A sites. This leads to higher m⁶A levels on BRCA1 mRNA, increased transcript degradation, impaired DNA repair, and heightened doxorubicin-induced DNA damage.
3. Could tadalafil be used clinically soon for breast cancer treatment?
Because tadalafil is already FDA-approved, repurposing it as a PRMT5 inhibitor is more feasible than developing a new drug from scratch. However, clinical trials are still needed to determine optimal dosing, patient selection, and safety in the context of chemotherapy. The preclinical data are extremely promising, but clinical validation remains essential.