Introduction: Why Reperfusion Injury Remains the Achilles’ Heel of Lung Transplantation
Despite remarkable advances in surgical technique, donor management, and perioperative care, lung transplantation continues to be burdened by a complication as old as the procedure itself: ischemia–reperfusion injury. Clinically, this manifests as primary graft dysfunction, a syndrome defined by hypoxemia, elevated pulmonary artery pressures, and noncardiogenic pulmonary edema within hours of transplantation. Its impact is immediate, profound, and often unforgiving.
Primary graft dysfunction does not merely complicate the early postoperative course. It sets the stage for longer intensive care stays, increased susceptibility to infection, higher rates of acute rejection, and reduced long-term graft survival. In this sense, ischemia–reperfusion injury is not an isolated event but a biological cascade whose consequences echo throughout the lifespan of the transplanted lung.
The persistence of this problem reflects a fundamental challenge: reperfusion injury is not driven by a single pathway. It represents the convergence of endothelial dysfunction, oxidative stress, inflammatory activation, and dysregulated vascular tone. Effective protection therefore requires interventions that act upstream, stabilizing cellular signaling before injury unfolds. Pharmacological preconditioning using long-acting phosphodiesterase inhibition emerges as one such strategy, grounded in vascular biology rather than surgical timing.
The Pathophysiology of Lung Ischemia–Reperfusion Injury
The lung is uniquely vulnerable to ischemia–reperfusion injury. Unlike solid organs with high metabolic reserve, pulmonary tissue depends on continuous perfusion to maintain endothelial integrity and gas exchange. Prolonged cold ischemia disrupts cellular homeostasis, but it is the moment of reperfusion that delivers the most damage.
Upon reperfusion, a sudden influx of oxygen into metabolically primed tissue generates reactive oxygen species. These free radicals damage cell membranes, disrupt mitochondrial function, and activate inflammatory cascades. Endothelial cells, in particular, lose their barrier function, leading to capillary leak and interstitial edema.
Simultaneously, pulmonary vascular resistance rises sharply. This increase is driven by endothelial dysfunction, impaired nitric oxide signaling, and enhanced vasoconstrictor activity. The result is elevated pulmonary artery pressure, right ventricular strain, and worsening ventilation–perfusion mismatch. In this environment, even structurally intact alveoli fail to oxygenate blood effectively.
Importantly, ischemia–reperfusion injury is not passive damage but an active biochemical process. Enzyme systems, intracellular signaling pathways, and transcriptional responses all contribute. This realization shifts therapeutic focus from reactive treatment to proactive modulation of molecular signaling before reperfusion begins.
Nitric Oxide, cGMP, and Pulmonary Vascular Homeostasis
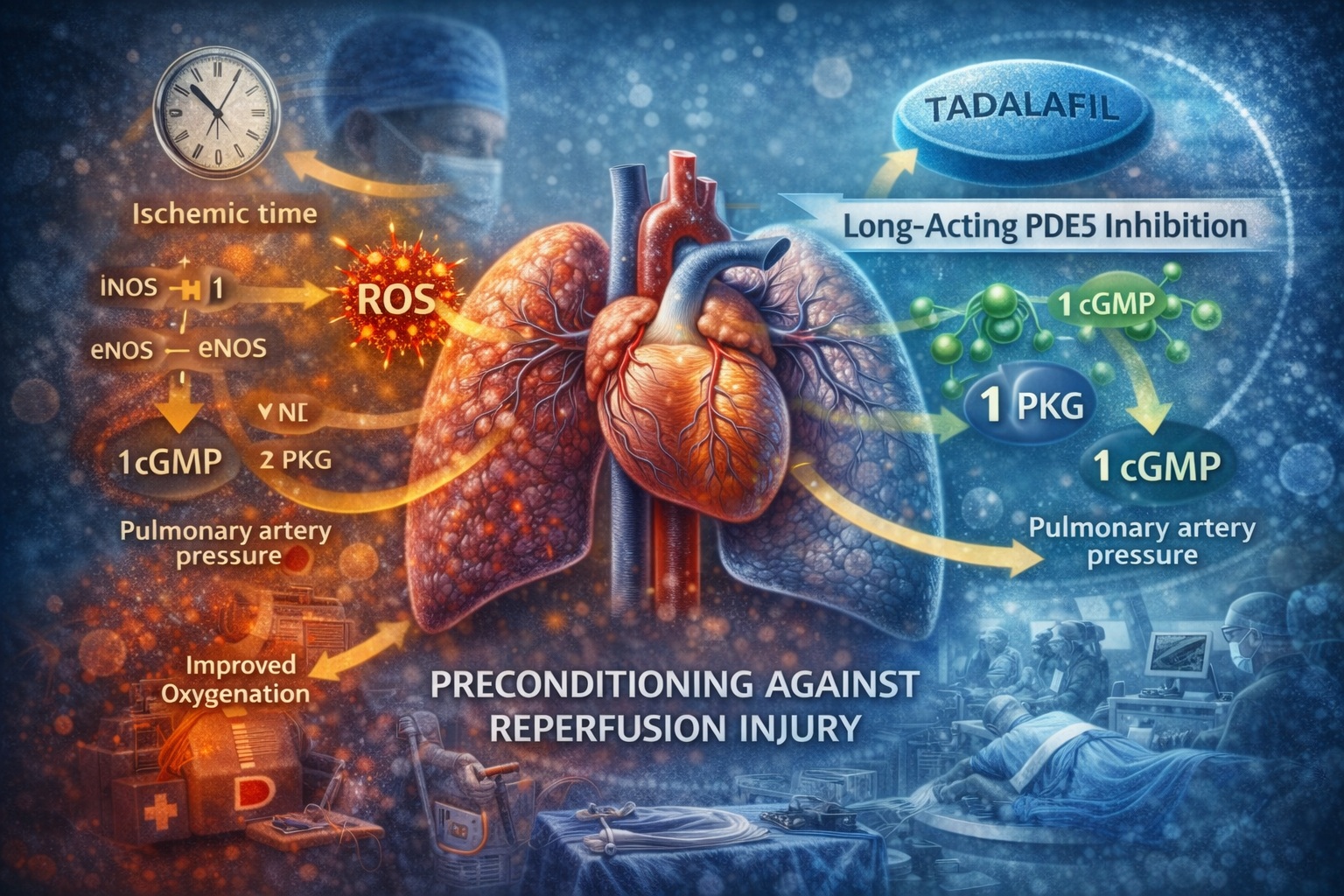
At the center of pulmonary vascular regulation lies the nitric oxide–cyclic guanosine monophosphate pathway. Under physiological conditions, endothelial nitric oxide synthase produces nitric oxide, which diffuses into adjacent smooth muscle cells. There, nitric oxide activates guanylate cyclase, increasing intracellular cGMP and triggering smooth muscle relaxation.
This pathway serves multiple protective functions in the lung. It maintains low pulmonary vascular resistance, preserves endothelial barrier integrity, inhibits platelet aggregation, and suppresses leukocyte adhesion. In essence, nitric oxide signaling keeps the pulmonary circulation both compliant and quiescent.
Ischemia–reperfusion injury disrupts this system at multiple levels. Nitric oxide bioavailability decreases, cGMP levels fall, and downstream signaling via protein kinase G is attenuated. At the same time, inducible nitric oxide synthase becomes upregulated, generating excessive and poorly regulated nitric oxide that paradoxically contributes to oxidative stress rather than protection.
The imbalance between protective endothelial nitric oxide signaling and injurious oxidative pathways represents a critical inflection point in reperfusion injury. Therapeutic strategies that preserve cGMP signaling offer a means of restoring this balance.
Phosphodiesterase-5: A Strategic Target in Reperfusion Injury
Phosphodiesterase enzymes regulate intracellular cyclic nucleotide levels by catalyzing their breakdown. Among these, phosphodiesterase type 5 plays a dominant role in degrading cGMP within vascular smooth muscle and endothelial cells. Increased PDE5 activity directly reduces cGMP availability, undermining nitric oxide–mediated protection.
Experimental evidence demonstrates that PDE5 activity rises during early reperfusion. This increase coincides with declining cGMP levels, reduced protein kinase G activation, worsening pulmonary hypertension, and impaired oxygenation. In effect, PDE5 acts as a molecular amplifier of reperfusion injury.
Selective inhibition of PDE5 therefore represents a rational therapeutic target. By preventing cGMP degradation, PDE5 inhibitors sustain downstream signaling even in the face of reduced nitric oxide availability. This approach does not require supraphysiologic nitric oxide production; it simply preserves the signaling that remains.
What makes PDE5 inhibition particularly attractive is its specificity. Unlike broader phosphodiesterase inhibitors, PDE5-selective agents focus on vascular signaling without globally disrupting cyclic nucleotide metabolism.
Tadalafil as a Long-Acting Preconditioning Agent
Among available PDE5 inhibitors, tadalafil possesses pharmacokinetic properties uniquely suited for donor preconditioning. Its long half-life allows sustained enzyme inhibition long after administration, and its oral bioavailability makes it practical in real-world donor management scenarios.
In experimental models, oral tadalafil administered hours before lung harvest achieves active inhibition of PDE5 within lung tissue at the time of reperfusion. This confirms that systemic administration can effectively precondition pulmonary vasculature without direct intrapulmonary delivery.
The extended duration of action distinguishes tadalafil from shorter-acting agents. In the context of organ transplantation, where timing is often unpredictable, a drug that maintains biological activity across prolonged ischemic intervals offers a clear advantage. Donor pretreatment can occur well before procurement without the need for continuous infusion or intraoperative manipulation.
This concept reframes PDE5 inhibitors from perioperative adjuncts to strategic preconditioning agents capable of reshaping the biochemical environment of the graft before injury occurs.
Functional Outcomes: Improved Oxygenation and Hemodynamics
Experimental lung transplantation models demonstrate that donor pretreatment with long-acting PDE5 inhibition yields meaningful functional benefits during reperfusion. Lungs exposed to prolonged cold ischemia typically show progressive declines in oxygenation upon reperfusion. In contrast, preconditioned lungs maintain higher arterial oxygen levels throughout the reperfusion period.
Hemodynamic stability follows a similar pattern. Mean pulmonary artery pressures rise sharply during reperfusion in untreated grafts, reflecting vasoconstriction and microvascular dysfunction. PDE5-inhibited lungs exhibit significantly lower pulmonary artery pressures, indicating preserved vasodilatory capacity and reduced vascular resistance.
These functional improvements are not transient artifacts. They persist across the reperfusion window and correlate with biochemical markers of preserved signaling. Notably, reductions in pulmonary edema are modest, suggesting that the primary benefit lies in vascular tone regulation and gas exchange rather than fluid balance alone.
From a clinical perspective, these findings are compelling. Improved oxygenation and lower pulmonary pressures translate directly into reduced ventilatory support, lower right ventricular strain, and a smoother postoperative course.
Molecular Evidence: Preserving cGMP and Protein Kinase G Signaling
Biochemical analysis confirms that the functional benefits of PDE5 inhibition are rooted in preserved intracellular signaling. Lung tissue exposed to tadalafil demonstrates reduced PDE5 activity during reperfusion, higher cGMP concentrations, and sustained activation of protein kinase G.
Protein kinase G serves as a central mediator of cGMP signaling, phosphorylating downstream targets that regulate vascular tone, mitochondrial stability, and cellular survival. Its preservation suggests that PDE5 inhibition stabilizes multiple protective pathways simultaneously.
Importantly, these effects are observed specifically during reperfusion rather than after cold storage alone. This distinction underscores the dynamic nature of reperfusion injury and highlights PDE5 activation as a reperfusion-specific event rather than a passive consequence of ischemia.
By maintaining cGMP–PKG signaling, PDE5 inhibition interrupts the feed-forward cycle of vasoconstriction, oxidative stress, and endothelial dysfunction that defines early graft injury.
Oxidative Stress and the Balance of Nitric Oxide Synthases
Reactive oxygen species are central mediators of reperfusion injury. Excessive ROS generation damages cellular structures and amplifies inflammatory responses. In experimental models, PDE5 inhibition significantly reduces ROS levels during reperfusion, indicating a protective effect at the oxidative level.
This reduction appears closely linked to nitric oxide synthase regulation. Endothelial nitric oxide synthase activity is preserved in preconditioned lungs, supporting controlled nitric oxide production and vascular protection. Conversely, inducible nitric oxide synthase activity is attenuated, limiting uncontrolled nitric oxide release and secondary oxidative damage.
This differential regulation is critical. Endothelial nitric oxide synthase supports coupling between nitric oxide and cGMP signaling, whereas inducible nitric oxide synthase often contributes to nitric oxide uncoupling and ROS generation. PDE5 inhibition appears to favor the former while suppressing the latter.
In practical terms, this means that PDE5 inhibition does not simply enhance nitric oxide signaling indiscriminately. It refines it, preserving physiological signaling while limiting pathological amplification.
Donor Preconditioning: A Shift in Transplant Strategy
Most interventions aimed at reducing ischemia–reperfusion injury focus on the recipient or the preservation solution. Donor preconditioning represents a conceptual shift, recognizing that the biological state of the graft before procurement influences outcomes after implantation.
Oral PDE5 inhibition fits naturally into this paradigm. It is noninvasive, easily administered, and compatible with the logistical realities of organ donation. Importantly, it acts systemically, conditioning the lung vasculature without the need for specialized equipment or intraoperative modification.
This approach may expand the donor pool by improving tolerance to prolonged ischemic times and marginal donor characteristics. In an era of organ scarcity, strategies that safely extend donor usability carry significant ethical and clinical weight.
Limitations and Future Directions
As with all experimental models, limitations must be acknowledged. Ex vivo perfusion systems, while highly controlled, cannot fully replicate the complexity of in vivo transplantation. Immune responses, neurohumoral influences, and long-term remodeling are not captured.
The durability of PDE5-mediated protection beyond early reperfusion remains an open question. While early functional improvements are clear, long-term effects on graft survival and chronic rejection require further investigation.
Future research should explore optimal dosing strategies, potential synergy with other preconditioning agents, and translation into clinical trials. Investigating PDE5 inhibition in combination with anti-inflammatory or antioxidant therapies may further enhance protection.
Conclusion
Ischemia–reperfusion injury remains a central obstacle in lung transplantation, driven by complex disruptions in vascular signaling, oxidative balance, and endothelial function. Long-acting phosphodiesterase-5 inhibition offers a biologically sound and practically feasible strategy for donor preconditioning.
By preserving cGMP signaling, stabilizing protein kinase G activity, reducing oxidative stress, and maintaining endothelial nitric oxide balance, PDE5 inhibition addresses multiple injury pathways simultaneously. Experimental evidence demonstrates meaningful improvements in oxygenation and pulmonary hemodynamics during reperfusion.
In a field where incremental advances can have life-saving consequences, pharmacological preconditioning with long-acting PDE5 inhibitors represents a promising step forward—one grounded not in surgical innovation, but in molecular precision.
FAQ
1. Why is reperfusion more damaging than ischemia itself?
Reperfusion introduces oxygen into metabolically primed tissue, triggering reactive oxygen species generation, endothelial dysfunction, and inflammatory activation that exceed ischemic injury alone.
2. What makes tadalafil suitable for donor preconditioning?
Its long half-life, oral bioavailability, and sustained PDE5 inhibition allow effective conditioning across unpredictable ischemic intervals.
3. Can this strategy be applied clinically?
The approach is biologically plausible and logistically feasible, but clinical trials are needed to confirm safety, optimal dosing, and long-term benefit in human lung transplantation.