
Introduction
Hepatocellular carcinoma (HCC) represents one of the most formidable challenges in oncology. Despite progress in surgery, local ablation, and systemic therapies, the disease continues to exact a high toll in terms of incidence and mortality worldwide. The emergence of epigenetic drugs, including bromodomain and extraterminal (BET) inhibitors, has sparked significant interest due to their ability to modulate oncogenic transcriptional programs. However, enthusiasm has been dampened by the sobering reality that BET inhibitor monotherapy shows only modest or negligible benefits in preclinical HCC models.
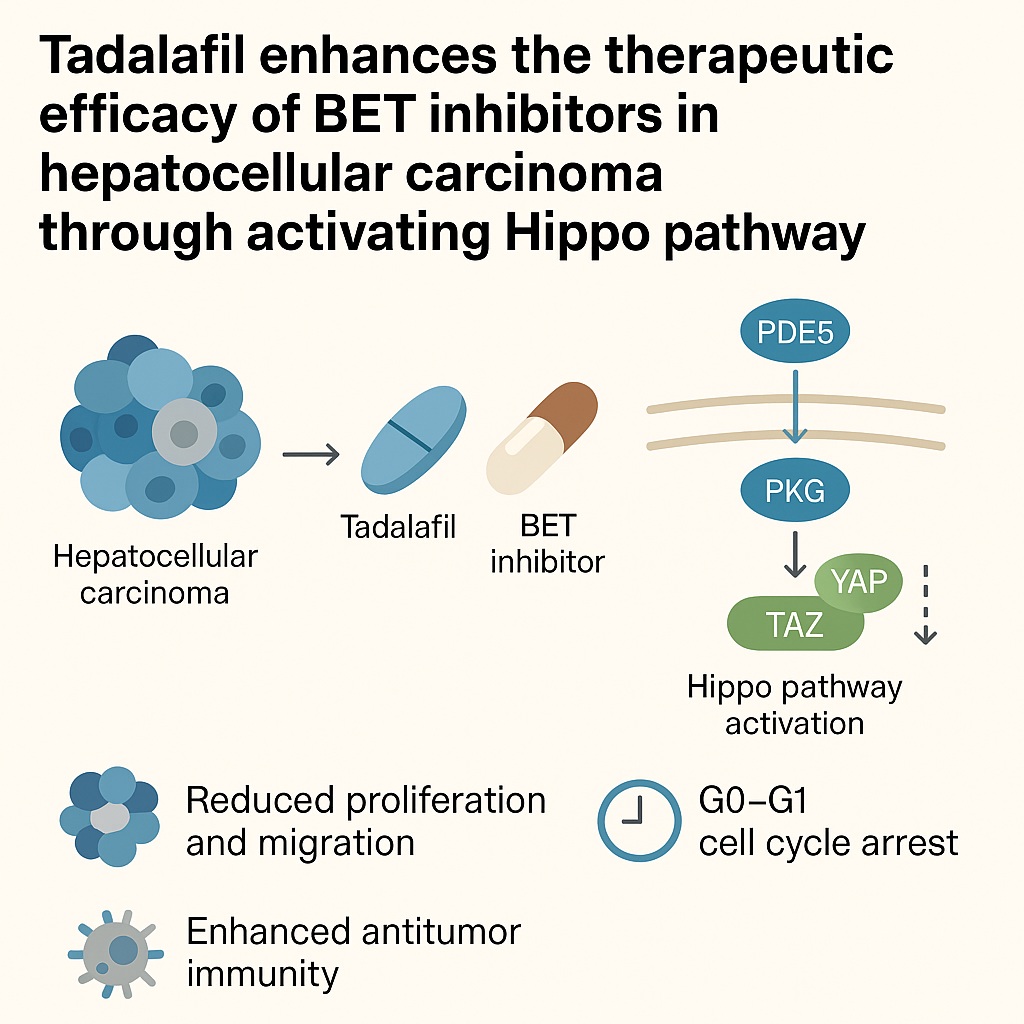
The recent exploration of combination regimens has reignited hope. One particularly intriguing development is the finding that tadalafil, a widely prescribed phosphodiesterase type 5 (PDE5) inhibitor, can potentiate the effects of BET inhibitors against HCC. This combination leverages the Hippo signaling pathway—a key regulatory axis of cell growth and apoptosis—to overcome resistance mechanisms that limit BET inhibitor efficacy. The repurposing of tadalafil, a drug better known for its roles in erectile dysfunction and pulmonary hypertension, into the oncology domain underscores the creativity of modern translational research.
This article provides a detailed analysis of the mechanistic basis, preclinical evidence, and potential clinical implications of combining tadalafil with BET inhibitors in HCC. Drawing upon recent experimental findings, it dissects the roles of YAP/TAZ transcriptional coactivators, the PDE5/PKG/Hippo/YAP/TAZ signaling axis, and the broader landscape of tumor immunity in shaping this therapeutic strategy.
BET Inhibitors in HCC: Promise and Limitations
BET proteins (BRD2, BRD3, BRD4, and BRDT) act as chromatin readers, recognizing acetylated lysines on histone tails and facilitating transcription of genes crucial for oncogenesis, including MYC. Pharmacological BET inhibition, exemplified by the small molecule JQ-1, displaces BET proteins from chromatin, leading to transcriptional silencing of oncogenes.
In vitro, BET inhibitors deliver measurable anticancer activity. HCC cell lines such as HepG2 and Huh7 exhibit reduced proliferation, cell cycle arrest, and even signs of apoptosis when exposed to JQ-1. However, when the same inhibitors are tested in vivo using oncogene-driven HCC mouse models, the outcomes have been disappointing. Tumor progression continues largely unabated, survival improvements are marginal, and resistance develops quickly.
The discrepancy between in vitro and in vivo outcomes illustrates the complex biology of HCC. Tumor heterogeneity, compensatory signaling pathways, and an immunosuppressive microenvironment all conspire to blunt the effects of single-agent BET blockade. These realities have shifted the focus toward rational combinations that can neutralize resistance drivers and restore therapeutic potency.
YAP/TAZ: Molecular Gatekeepers of Resistance
Central to the story is the Hippo signaling pathway and its downstream transcriptional coactivators, Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ). Normally, the Hippo pathway restrains cell proliferation by phosphorylating YAP and TAZ, thereby sequestering them in the cytoplasm and targeting them for degradation. When Hippo signaling is dysregulated, YAP and TAZ translocate to the nucleus, where they drive expression of genes that promote proliferation, survival, and stemness.
In HCC, elevated YAP/TAZ activity is strongly correlated with aggressive behavior, metastasis, and poor clinical outcomes. Importantly, YAP/TAZ expression confers resistance to multiple anticancer agents, including BET inhibitors. Tumor cells with active YAP/TAZ programs remain proliferative despite BET blockade, essentially bypassing the intended transcriptional silencing.
Silencing YAP or TAZ with siRNA markedly sensitizes HCC cells to JQ-1. Conversely, forced overexpression of these proteins reduces drug sensitivity, confirming their causal role in resistance. Interestingly, TAZ appears to exert a stronger effect than YAP in this context, suggesting nuanced, nonredundant roles for these paralogs.
The PDE5/PKG/Hippo Axis: An Unexpected Vulnerability
Direct pharmacological inhibitors of YAP/TAZ do not exist. However, molecular analysis has revealed that their regulation is intimately connected with PDE5, the enzyme responsible for degrading cyclic guanosine monophosphate (cGMP). High PDE5 expression correlates with elevated YAP/TAZ activity in HCC, establishing a link between PDE5 signaling and Hippo pathway dysregulation.
PDE5 inhibitors such as tadalafil elevate cGMP levels, thereby activating protein kinase G (PKG). Activated PKG, in turn, phosphorylates components of the Hippo cascade, including MST and LATS kinases. This phosphorylation restores Hippo pathway activity, promoting YAP/TAZ degradation and reducing their transcriptional activity.
In HepG2 cells treated with tadalafil, RNA sequencing revealed downregulation of YAP1 and WWTR1 (TAZ) expression. Western blot analysis confirmed reduced YAP/TAZ protein levels, consistent with Hippo pathway activation. Thus, tadalafil indirectly accomplishes what no direct YAP/TAZ inhibitor can: it silences their oncogenic output by restoring upstream regulation.
Tadalafil Meets BET Inhibition: A Synergistic Partnership
Armed with this mechanistic insight, investigators tested the hypothesis that tadalafil could sensitize HCC cells to BET inhibition. The results were striking.
When HepG2 cells were treated with tadalafil plus JQ-1, several synergistic effects emerged:
- Suppression of YAP/TAZ expression: The combination led to greater reductions than either agent alone.
- Inhibition of proliferation and colony formation: Cells exposed to the combination exhibited profound growth arrest.
- Impaired migration and invasion: The metastatic potential of HCC cells was sharply curtailed.
- Cell cycle arrest: The combined regimen induced a robust G0-G1 arrest, effectively halting cell division.
These findings highlight the elegance of combination therapy. By dismantling YAP/TAZ-driven resistance, tadalafil restores the ability of BET inhibitors to exert full antitumor activity.
Antitumor Immunity: A Hidden Bonus
HCC progression is not merely a story of tumor-intrinsic signaling. The immune microenvironment plays a pivotal role, often tilting the balance in favor of immune escape. BET inhibitors themselves can modulate immune checkpoints, including the PD-1/PD-L1 axis.
In preclinical models, JQ-1 reduces interferon-γ-induced PD-L1 expression in HCC cells, although this effect is inconsistent across cell lines. Remarkably, when combined with tadalafil, the downregulation of PD-L1 becomes more pronounced, suggesting an immunomodulatory synergy.
Flow cytometric analyses of HCC-bearing mice revealed that combination therapy increased the proportion of activated CD8+ cytotoxic T cells while reducing immunosuppressive populations such as myeloid-derived suppressor cells (MDSCs). These changes translated into enhanced antitumor immunity, an effect that may further amplify the direct cytostatic and cytotoxic effects of the drug regimen.
In Vivo Evidence: Tumor Suppression and Survival Gains
The ultimate test of any therapy lies in animal models that recapitulate the complexity of human disease. In c-Myc/N-Ras-driven transgenic HCC mice, neither tadalafil nor JQ-1 monotherapy significantly altered tumor burden. Liver sizes, tumor nodules, and histological features remained largely unchanged compared with controls.
The picture changed dramatically with combination therapy. Mice receiving both drugs exhibited:
- Reduced tumor progression: Fewer and smaller nodules, lower liver/body weight ratios, and reduced proliferation markers such as Ki-67.
- Improved survival: Median survival extended from ~70 days in controls to over 112 days in the combination group.
- Enhanced immune activation: As noted, activated CD8+ T cells were more abundant.
These results underscore that the partnership between tadalafil and BET inhibition is not merely additive—it is synergistic, producing therapeutic benefits unattainable with either agent alone.
Broader Implications and Future Directions
The repurposing of tadalafil for oncology exemplifies the value of exploring unexpected intersections between pharmacology and cancer biology. Several implications deserve emphasis:
- Clinical translatability: Tadalafil is FDA-approved, widely available, and has a well-characterized safety profile. This lowers barriers to clinical testing in HCC.
- Biomarker potential: Elevated YAP/TAZ or PDE5 expression may identify patients most likely to benefit from the combination.
- Beyond HCC: Given the involvement of YAP/TAZ in resistance across multiple cancers, this strategy could extend to other malignancies, including lung adenocarcinoma and breast cancer.
Challenges remain, of course. Preclinical models are not perfect surrogates for human disease. Dosing strategies must be optimized, toxicity evaluated, and potential interactions with existing HCC therapies such as sorafenib or immunotherapy considered. Yet the conceptual leap is significant: by harnessing a common urological drug, oncologists may unlock a new weapon against one of the deadliest cancers.
Conclusion
The convergence of BET inhibition and PDE5 blockade represents a bold step toward overcoming therapeutic resistance in hepatocellular carcinoma. By targeting the Hippo pathway and silencing YAP/TAZ, tadalafil restores and amplifies the anticancer potential of BET inhibitors. The preclinical evidence is compelling: combination therapy suppresses proliferation, impedes metastasis, enhances antitumor immunity, and prolongs survival in animal models.
What began as a strategy to manage erectile dysfunction may now help combat a lethal malignancy. If clinical trials confirm these findings, tadalafil could become a cornerstone in the evolving armamentarium against HCC, proving once again that innovation often emerges from the most unexpected intersections of medicine.
FAQ
1. Why do BET inhibitors alone fail to control hepatocellular carcinoma?
Although BET inhibitors effectively suppress HCC cell growth in vitro, they encounter resistance in vivo due to compensatory signaling pathways, particularly activation of YAP/TAZ. This resistance undermines their ability to control tumor progression in animal models and likely in patients.
2. How does tadalafil enhance the efficacy of BET inhibitors?
Tadalafil inhibits PDE5, raising intracellular cGMP levels and activating PKG. This activation restores Hippo pathway activity, leading to phosphorylation and degradation of YAP/TAZ. By silencing these resistance mediators, tadalafil sensitizes tumor cells to BET inhibition.
3. Could tadalafil be repurposed for cancers beyond HCC?
Yes. Since YAP/TAZ overexpression drives resistance in multiple malignancies, the combination of PDE5 inhibitors with BET inhibitors—or even with other targeted drugs—holds promise for a variety of cancers, pending further preclinical and clinical validation.