
Pulmonary arterial hypertension (PAH) remains one of the most challenging disorders in cardiovascular medicine. Despite considerable therapeutic progress over the past two decades, the disease continues to carry significant morbidity and mortality. Modern pharmacology has transformed PAH from an almost uniformly fatal condition into a chronic disease that can be managed for years, sometimes decades. Yet clinicians and researchers agree that current therapies are still imperfect.
Most available treatments target well-known signaling pathways involved in pulmonary vascular tone and remodeling. These include the endothelin pathway, the nitric oxide–cyclic GMP pathway, and the prostacyclin pathway. Drugs such as endothelin receptor antagonists, prostacyclin analogues, and phosphodiesterase type-5 inhibitors—including tadalafil—have improved outcomes significantly. However, many patients continue to experience progressive disease despite combination therapy.
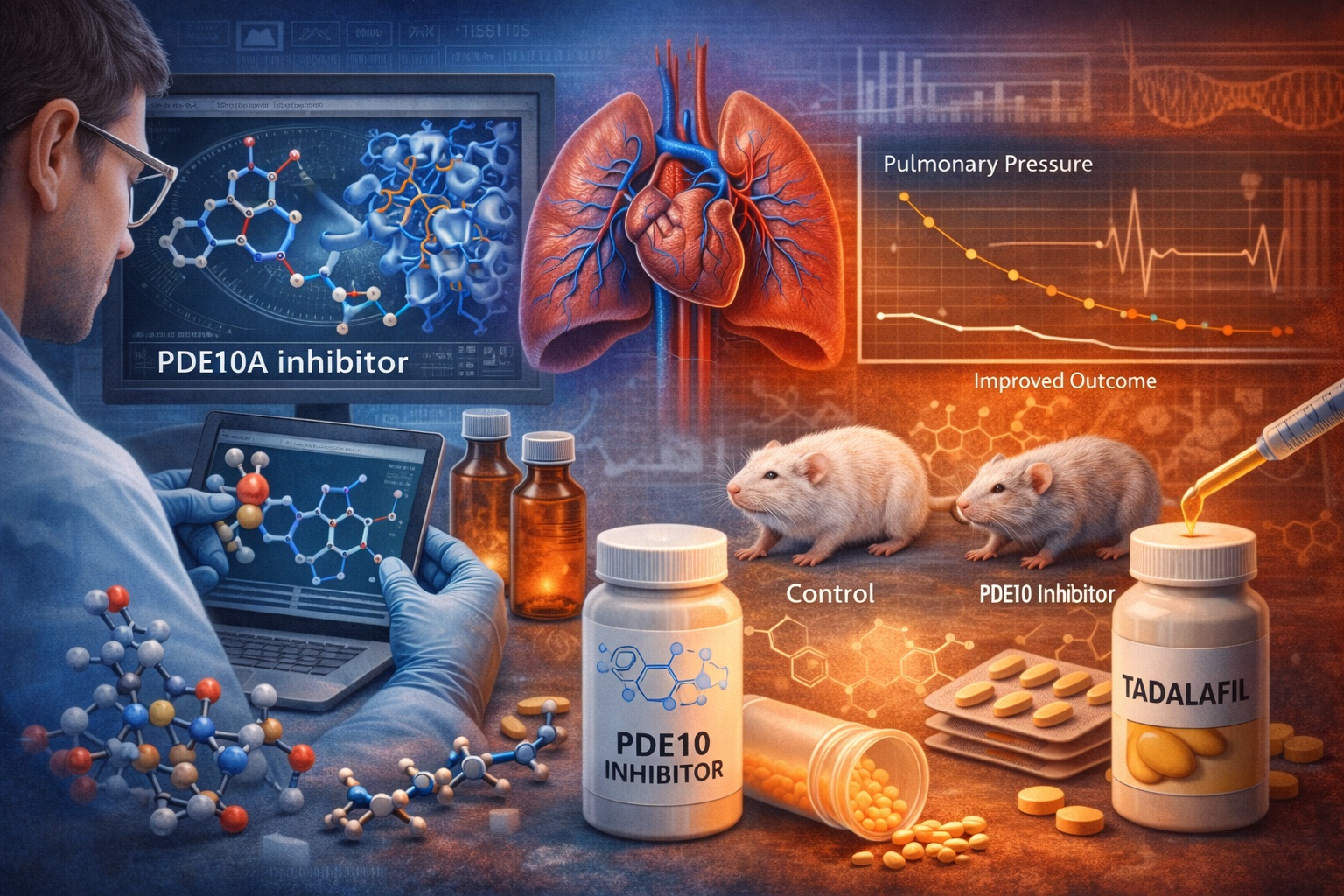
The search for novel pharmacological targets therefore remains an active field of research. Among the most promising areas of investigation is the role of phosphodiesterase 10A (PDE10A) in pulmonary vascular regulation. Recent drug discovery efforts have focused on the development of selective inhibitors of this enzyme, particularly compounds based on benzimidazole scaffolds with improved pharmacokinetic properties.
The discovery of orally available, highly selective PDE10 inhibitors represents a significant step forward in the search for next-generation PAH therapies. These compounds combine advanced medicinal chemistry, structural biology, and pharmacological evaluation to produce molecules with favorable solubility, metabolic stability, and therapeutic potential.
This article explores the scientific rationale behind PDE10 inhibition, the development of benzimidazole-based inhibitors, and their potential role in the evolving treatment landscape of pulmonary arterial hypertension. The story is not merely one of chemical innovation—it is also a demonstration of how modern drug design can reshape our therapeutic strategies.
Understanding Pulmonary Arterial Hypertension: A Disease of Vascular Imbalance
Pulmonary arterial hypertension is defined by progressive elevation of pulmonary arterial pressure caused by structural and functional changes in the pulmonary vasculature. Over time, these changes increase pulmonary vascular resistance, forcing the right ventricle to work harder in order to maintain blood flow through the lungs.
Initially, the right ventricle adapts by hypertrophying and increasing contractility. However, this adaptation is ultimately limited. As pulmonary resistance continues to rise, the right ventricle begins to fail, leading to decreased cardiac output, systemic congestion, and ultimately life-threatening complications.
At the cellular level, PAH is characterized by a complex network of pathological processes. These include endothelial dysfunction, excessive smooth muscle proliferation, inflammation, and metabolic alterations. The pulmonary arteries gradually narrow as their walls thicken and lose their ability to relax appropriately.
A key feature of PAH is the disruption of signaling pathways that regulate vascular tone. Normally, pulmonary arteries maintain a balance between vasoconstrictor and vasodilator influences. In PAH, this balance is disturbed. Vasoconstrictive mediators become dominant, while protective signaling mechanisms weaken.
This imbalance explains why modern PAH therapies focus on restoring physiological signaling pathways. Drugs that enhance nitric oxide signaling or mimic prostacyclin activity can help relax pulmonary arteries and reduce vascular resistance. Likewise, endothelin receptor antagonists block the effects of powerful vasoconstrictor peptides.
Yet despite these advances, many patients remain symptomatic even with combination therapy. The persistence of vascular remodeling and abnormal signaling indicates that additional molecular targets must be explored. Among these emerging targets, phosphodiesterase enzymes have attracted considerable attention.
Phosphodiesterases and Vascular Signaling: Why PDE10 Matters
Phosphodiesterases (PDEs) are enzymes responsible for degrading cyclic nucleotides such as cyclic AMP (cAMP) and cyclic GMP (cGMP). These molecules serve as intracellular messengers that regulate numerous physiological processes, including vascular relaxation and smooth muscle contraction.
Within the pulmonary circulation, cyclic GMP plays a particularly important role. When nitric oxide stimulates soluble guanylate cyclase, intracellular cGMP levels rise, leading to relaxation of pulmonary arterial smooth muscle. This mechanism forms the basis of therapy with PDE5 inhibitors such as tadalafil, which prolong the action of cGMP by preventing its breakdown.
However, PDE5 is not the only enzyme capable of regulating cyclic nucleotide signaling. Multiple PDE isoforms exist throughout the body, each with distinct tissue distributions and biochemical properties. Among them, phosphodiesterase 10A (PDE10A) has emerged as a potentially important regulator of vascular signaling.
Originally studied in the central nervous system, PDE10A was known for its involvement in neuronal signaling pathways. More recent research has demonstrated that PDE10A is also expressed in peripheral tissues, including vascular smooth muscle cells.
By modulating intracellular levels of cyclic nucleotides, PDE10A may influence vascular tone and cellular proliferation. In pulmonary hypertension, where abnormal smooth muscle growth and impaired vasodilation are central features, inhibition of PDE10A could theoretically produce beneficial effects.
This insight has inspired medicinal chemists to explore PDE10A as a therapeutic target. If selective inhibitors could be developed with suitable pharmacokinetic properties, they might represent an entirely new class of pulmonary vasodilators.
Of course, identifying a promising molecular target is only the beginning. The real challenge lies in designing molecules capable of inhibiting the enzyme with high potency, selectivity, and favorable drug-like properties.
Medicinal Chemistry Breakthrough: Benzimidazole-Based PDE10 Inhibitors
The development of PDE10 inhibitors represents a classic example of rational drug design. Researchers began with previously identified PDE10 inhibitory compounds and sought to optimize their chemical structures to improve potency, selectivity, and pharmacokinetic behavior.
One particularly successful strategy involved the use of benzimidazole-based molecular scaffolds. Benzimidazole derivatives have long been recognized in medicinal chemistry for their structural versatility and biological activity. By modifying substituent groups around the benzimidazole core, chemists can fine-tune molecular interactions with specific enzyme binding sites.
The optimization process required several iterative steps. Researchers synthesized a series of compounds and evaluated their inhibitory activity against PDE10A using biochemical assays. Compounds demonstrating strong activity were then tested for selectivity against other phosphodiesterase isoforms.
Selectivity is critically important. Many PDE enzymes share structural similarities, and non-selective inhibition can produce unwanted side effects. For example, inhibition of PDE3 may influence cardiac contractility, while PDE4 inhibition can lead to gastrointestinal symptoms.
Through careful structural modifications, researchers were able to develop compounds with extremely high selectivity for PDE10A. One optimized molecule demonstrated nanomolar inhibitory potency along with remarkable selectivity over other PDE enzymes.
Beyond potency and selectivity, researchers also addressed practical drug development challenges such as solubility and metabolic stability. Poor solubility is a common obstacle in drug design because it limits oral absorption. Similarly, compounds that are rapidly metabolized in the liver may fail to achieve therapeutic plasma concentrations.
By modifying chemical substituents and analyzing structure–activity relationships, investigators produced compounds with significantly improved solubility and metabolic stability. These improvements allowed the molecules to be administered orally with favorable bioavailability.
In short, the resulting benzimidazole-based inhibitors combined three critical features: strong PDE10 inhibition, high selectivity, and drug-like pharmacokinetic properties.
Structural Biology and Rational Drug Design
One of the most powerful tools in modern drug discovery is structural biology. By determining the three-dimensional structure of a drug bound to its target enzyme, researchers can visualize the molecular interactions responsible for inhibitory activity.
In the case of PDE10 inhibitors, crystallographic analysis revealed how benzimidazole-based molecules bind within the enzyme’s active site. The structural data showed a highly specific arrangement of hydrogen bonds, hydrophobic contacts, and aromatic stacking interactions.
These interactions stabilize the inhibitor within the catalytic pocket of PDE10A, effectively blocking access to the enzyme’s natural substrates. The precision of these interactions explains the remarkable potency of the optimized compounds.
Structural analysis also provided insights into why the molecules exhibit strong selectivity for PDE10A compared with other phosphodiesterases. Subtle differences in amino acid residues surrounding the catalytic site create unique binding environments that favor specific molecular shapes.
Armed with this structural information, researchers could further refine compound design. Adjustments to functional groups allowed chemists to strengthen beneficial interactions while avoiding steric clashes with nearby residues.
This iterative process—combining biochemical testing with structural analysis—represents the essence of rational drug design. Rather than relying on trial and error, scientists can use molecular insight to guide the creation of increasingly effective compounds.
The resulting inhibitors are therefore not accidental discoveries. They are carefully engineered molecules shaped by a deep understanding of enzyme structure and function.
Pharmacokinetics and Therapeutic Potential
Even the most potent enzyme inhibitor is useless as a medicine if it cannot reach its target in the body. For this reason, pharmacokinetic evaluation is a crucial step in drug development.
Researchers assessed the absorption, distribution, metabolism, and elimination of optimized PDE10 inhibitors using animal models. These studies demonstrated encouraging results. The compounds exhibited improved metabolic stability and a favorable bioavailability profile when administered orally.
Bioavailability is particularly important for chronic diseases such as pulmonary arterial hypertension. Patients often require lifelong therapy, making oral administration far more practical than injectable medications.
In experimental models of pulmonary hypertension, treatment with optimized PDE10 inhibitors produced meaningful improvements in pulmonary vascular parameters. Remarkably, the therapeutic effects observed in animal studies were comparable to those produced by tadalafil, a well-established PDE5 inhibitor widely used in PAH management.
This comparison is noteworthy because tadalafil has become one of the cornerstone therapies for PAH. Demonstrating similar efficacy in experimental models suggests that PDE10 inhibition could represent a viable alternative or complementary therapeutic approach.
Of course, translating these findings into clinical practice will require extensive additional research, including safety evaluation and human clinical trials. Nevertheless, the early pharmacological results are encouraging and suggest that PDE10 inhibitors may eventually expand the therapeutic arsenal against PAH.
Integrating PDE10 Inhibitors into Future Treatment Strategies
The modern treatment of pulmonary arterial hypertension increasingly relies on combination therapy targeting multiple molecular pathways. Current guidelines often recommend initiating treatment with two or more agents affecting different signaling systems.
In this context, PDE10 inhibitors could provide an additional mechanism for restoring cyclic nucleotide balance in pulmonary vascular cells. Their activity may complement existing drugs such as endothelin receptor antagonists or PDE5 inhibitors.
One intriguing possibility is that PDE10 inhibition may enhance cyclic nucleotide signaling through pathways that are not fully addressed by current therapies. By influencing both cAMP and cGMP signaling cascades, PDE10 inhibitors may produce broader vascular effects than agents targeting a single pathway.
Future clinical research will determine whether these compounds are best used as monotherapy, combination therapy, or perhaps as second-line treatments for patients who do not respond adequately to existing medications.
Regardless of their final clinical role, the discovery of selective benzimidazole-based PDE10 inhibitors highlights an important principle in cardiovascular pharmacology: progress often emerges from the intersection of molecular biology, medicinal chemistry, and clinical insight.
And occasionally, the most promising breakthroughs come not from entirely new ideas, but from re-examining familiar biochemical pathways with fresh scientific tools.
FAQ
What are PDE10 inhibitors and why are they important for pulmonary hypertension?
PDE10 inhibitors block the phosphodiesterase 10A enzyme, which regulates intracellular cyclic nucleotide signaling. By inhibiting this enzyme, these compounds may improve pulmonary vascular relaxation and reduce pathological remodeling in pulmonary arterial hypertension.
How do PDE10 inhibitors compare with tadalafil?
Tadalafil is a PDE5 inhibitor that enhances nitric oxide–mediated vasodilation. Experimental studies suggest that certain PDE10 inhibitors may produce therapeutic effects comparable to tadalafil in animal models of pulmonary hypertension.
Are PDE10 inhibitors currently available for clinical use?
At present, PDE10 inhibitors for pulmonary arterial hypertension remain in the research stage. While early results are promising, further studies—including human clinical trials—are necessary before these compounds can become approved therapies.
